
10/03/2021 (2021-01-16)
Auteur(s) : Valère Lounnas Ph.D. et Dr Gérard Guillaume
[NdNM : Le chapitre 1 a été publié sur FranceSoir : Histoire de la Covid-19 – chapitre 1 (francesoir.fr)]
Histoire de la Covid-19 — Chapitre 1
Partie 1 — Le marché aux fruits de mer de Wuhan, un coupable peut-être tout désigné ?
Connaître avec exactitude le point de départ géographique (épicentre ou point zéro) d’une épidémie est primordial pour en identifier la source biologique (réservoir) et le mode de transmission à l’homme (vecteur). Cela permet ainsi non seulement d’éradiquer le foyer initial, ce qui est de toute évidence la première des mesures d’endiguement de l’épidémie, mais aussi de comprendre les mécanismes d’interaction de l’homme avec son environnement et les circonstances qui ont conduit à son déclenchement. Tout cela dans le but essentiel d’anticiper les résurgences possibles de l’épidémie et donc de faciliter son contrôle ultérieur.
Identifier le point de départ d’une épidémie se compare donc à une enquête policière et doit se résoudre avec la même minutie. De l’exactitude des premières constatations découle inéluctablement la réussite de l’enquête. Il est important, donc, que le plus grand nombre d’éléments potentiellement indicatifs des faits soient collectés et rassemblés le plus tôt possible. Dans les enquêtes policières il arrive parfois que l’on ne connaisse pas la scène du crime, le corps de la victime ayant été déplacé pour dissimuler le crime ou qu’il ne soit signé de façon trop évidente et ainsi empêcher toute enquête. Les enquêteurs doivent alors procéder à un long travail méthodique d’investigation et de recoupement de faits sur les circonstances de la disparition de la victime sans garantie d’aboutir.
Avant de rentrer dans le détail de l’analyse concernant l’épicentre exact de l’épidémie, il est utile de faire une digression importante pour rappeler que la presse internationale et française ont pris l’habitude de dénommer systématiquement le virus de la pandémie Covid-19 comme étant le « nouveau coronavirus » ou SARS-Cov2. Un acronyme anglais qui signifie en français : deuxième coronavirus du syndrome respiratoire aigu sévère, en référence à un coronavirus très similaire, le SARS-Cov, dont l’épidémie de 2002-2003 avait touché 30 pays et aurait pu devenir une pandémie mondiale si elle n’avait pas pu être jugulée à temps. Ce terme, mis à toutes les sauces rédactionnelles, véhicule de façon involontaire, mais évidente une information essentielle qui, comme nous le montrerons dans la suite de cet article, contredit le discours de nos gouvernants, en France et en Europe, qui s’acharnent à nous dire que l’on est dans un processus historique où on apprend tout d’un virus nouveau et de sa maladie associée.
Cela relève de la désinformation et de la manipulation de masse. Car s’il est vrai que la pandémie a atteint une ampleur mondiale au moins mille fois plus considérable que celle de 2002-2003, qui n’avait fait que 776 morts pour 8000 personnes ayant présenté des symptômes (taux de létalité 9,6 %), il n’en reste pas moins que cette maladie était causée par un virus très proche du SARS-Cov2. Elle se présentait également sous la même forme de pneumonie atypique avec des conséquences respiratoires drastiques, incontrôlables, menant une grande proportion de patients atteints à des situations de mise sous assistance respiratoire mécanique et coma artificiel pendant une période de 10 jours à 3 semaines ! Une des différences essentielles avec le SARS-Cov2 est que la période d’incubation était beaucoup plus courte et la progression vers l’atteinte respiratoire plus rapide. Nous voyons donc que l’affirmation d’une maladie entièrement nouvelle ne tient pas, mais qu’elle permet, par contre, d’éviter un débat qui exposerait les raisons pour lesquelles aucune mesure n’a été prise pour prévenir la résurgence d’une telle maladie, ni aucune mesure prise pour anticiper l’arrivée massive de patients en réanimation. Cela a permis également de porter le débat médiatique dans les premiers jours de l’épidémie en France sur la fausse question de l’identification du réservoir animal qui est, comme nous le verrons par la suite, sans contestation possible la chauve-souris.
La seule question étant de savoir comment le SARS-Cov2 a franchi la barrière des espèces, soit directement, soit via un hôte intermédiaire, ou soit encore à la suite d’une manipulation de laboratoire. Pour le SARS-Cov il avait été établi que l’hôte intermédiaire, entre la chauve-souris et l’homme, était la civette palmiste, un gibier d’élevage vendu vivant sur le marché aux fruits de mer de Canton ! Il semblerait que là aussi il n’est pas dans l’intérêt de nos gouvernants que ces deux épidémies soient mises en rapport l’une avec l’autre sur le plan de l’infectiologie. Deux types de questions se posent immédiatement :
- À l’aune de ce que nous expliquons au premier paragraphe, sur la prévention des résurgences, les citoyens du monde sont en droit de demander à l’OMS pourquoi, après qu’il ait été établi scientifiquement que la source de la pandémie hautement mortelle de 2003 était le marché aux fruits de mer de Canton, la Chine n’a pas fermé ses autres marchés aux animaux sauvages comme celui de Wuhan que les autorités chinoises ont très rapidement présenté comme l’origine de l’épidémie locale de Covid-19 ? Il y a là une des défaillances notoires de l’OMS actuellement contrôlée par la Chine avec le mandat de son président actuel, le Dr Tedros Adhanom Ghebreyesus, élu Directeur général de l’OMS pour un mandat de 5 ans, en mai 2017.
- Outre la mise en cause de leur responsabilité par leur incurie, les gouvernants risqueraient de devoir répondre en détail sur les raisons des très grandes craintes qu’ils affichent concernant différents aspects fantasmés de l’épidémie comme celle de ne pas la voir disparaître, celle que l’on puisse développer le virus une seconde fois, ou celle qu’un vaccin ne puisse être mis au point. Pourquoi avoir fait preuve de tant d’incurie auparavant et à présent de tant de peur ? Il y a là semble-t-il une contradiction profonde. La collusion en France entre nos gouvernants et l’industrie pharmaceutique est bien connue du grand public à présent, mais ne suffit pas à expliquer une telle peur. Il s’agirait peut-être plutôt d’une crainte fondée sur le fait que la répétition de pandémies de coronavirus d’une telle nature, voire même peut-être pire, est à redouter dans les années à venir, pour des raisons que les grands médias classiques sont incapables d’expliquer ou ont pour ordre de ne pas exposer au public.
L’étude systématique des coronavirus issus de chauves-souris par les chercheurs militaires et universitaires chinois ainsi qu’une réalité technologique certaine, qui permet la manipulation et la création de virus Covid synthétiques, font craindre l’émergence potentielle de pandémies accidentelles causées par des virus qui ont été à un moment donné manipulés par l’homme qui finissent par échapper à son contrôle et réinfecter son environnement, directement ou indirectement.
De tels accidents, dus à des expérimentations biologiques, ont déjà eu lieu dont le plus célèbre est celui du virus de la fièvre hémorragique de Marbourg (ville d’Allemagne) qui avait contaminé 31 chercheurs et laborantins de la société Behring dont 7 périrent. Ils travaillaient à l’élaboration de vaccins à partir de cellules rénales de singes verts qui avaient été récemment importés d’Ouganda. À noter d’ailleurs que le réservoir naturel de ce virus si mortel de Marbourg, qui se rapproche de celui d’Ebola, a été finalement identifié en 2007 ; il s’agit une fois de plus de la chauve-souris comme pour celui d’Ebola ! Cette découverte a d’ailleurs coûté la vie à quelques explorateurs imprudents qui se sont aventurés dans la fameuse grotte de Kitum au Kenya (ndlr : modifié le 30/12 d’Ouganda en Kenya) y contractant la maladie mortelle. Nous soulignons ici la dangerosité des virus portés par les chauves-souris et il est bien connu à ce titre qu’il faut éviter de pénétrer leur habitat qui souvent est une grotte reculée. Ces animaux ancestraux sont le réservoir naturel des maladies les plus terribles. En France, par exemple, certaines chauves-souris sont porteuses de lyssavirus (un certain type de virus de la rage). La destruction de l’habitat de ces animaux ou bien leur prélèvement pour en faire des animaux de laboratoire, comme cela a été fait sciemment en Chine, constitue indubitablement une première étape d’un processus possible de franchissement de la barrière des espèces, dû à l’homme.
Suite à l’accident de Marbourg, on a construit des laboratoires biologiques de très haute sécurité (niveau P4) pour y conduire des manipulations de virus. Deux laboratoires P4 existent en Chine tels que celui de Wuhan, le premier construit, qui rappelons-le est une réplique de celui de Pasteur-Mérieux de Lyon Gerland, construit sous l’égide de la France et mis en service officiellement en 2017 (voir l’article publié par FranceSoir à la suite de celui-ci). Cependant, ce niveau de sécurité n’exclut pas la possibilité qu’un virus mortel (naturel ou manipulé) puisse s’en échapper accidentellement.
La seconde partie de cet article, ainsi ceux qui suivront, n’a pas pour but uniquement de retracer la chronologie des faits que le lecteur peut retrouver dans un très long dossier du journal Le Monde, mais d’en faire une synthèse et une analyse critique qui permet de faire ressortir les incohérences d’une présentation des faits trop simpliste et réductrice dans le discours de nos gouvernants.
Partie 2 — Fermeture des marchés aux fruits de mer de Wuhan, destruction de preuves et désinformation
Fermeture des marchés aux fruits de mer
Le 30 décembre 2019, les autorités de Wuhan font référence à l’épidémie pour la première fois sur internet dans une circulaire interne de la Commission Municipale de Santé (Wuhan Municipal Health). Un cas de pneumonie a été détecté chez une femme le 16 décembre 2019, suffisamment atypique pour que les autorités locales s’en préoccupent. La circulaire indique que toutes les unités médicales de Wuhan sont tenues de signaler à l’administration médicale et sanitaire, les cas similaires de pneumonie de cause inconnue.

« Dès le 30 décembre 2019, Li Wenliang, médecin de 34 ans de l’hôpital central de Wuhan, avait attiré l’attention de ses collègues sur le fait que sept personnes travaillant sur le marché aux animaux sauvages de la capitale de la province du Hubei étaient hospitalisées et semblaient avoir contracté un virus proche du syndrome respiratoire aigu sévère (SRAS). » (Le Monde).
Cette déclaration libre a précipité incontestablement l’annonce publique le jour suivant, le 31 décembre, par la Commission de Santé de Wuhan Municipal de plusieurs cas de pneumonie déclarés, liés au marché des fruits de mer de Huanan à Wuhan.
Moins fiable et donc sujette à caution, puisqu’elle n’est pas vérifiable, une seconde information démontrant que les autorités chinoises locales, et donc forcément nationales, savaient la dangerosité réelle du virus dès le 27 décembre. Le laboratoire VisionLabs, de Guangzhou, aurait séquencé le virus responsable de la pneumonie, démontrant qu’il s’apparentait au SARS-CoV, apparu en octobre 2002. Le laboratoire aurait reçu ordre de détruire ces échantillons (par qui ?) dans un contexte de tensions entre Pékin et les autorités locales de Wuhan selon Caixin, un média de réputation progressiste dont le siège se trouve pourtant à Pékin sous le regard du gouvernement central. Cela est donc, disons-le, peu crédible et ressemble plutôt à une vision occidentalisée de rapports conflictuels entre autorités locales et centrales. Ce genre de conflit n’existe certainement pas dans le régime chinois actuel. L’intérêt de la parution d’une telle information est, d’une certaine façon, la déresponsabilisation de gouvernement central chinois qui peut ainsi prétexter un défaut de communication avec les autorités locales pour couvrir un certain retard, durant la deuxième moitié du mois de décembre, à alerter l’OMS et le monde.
En tout état de cause, le 1er janvier 2020, les autorités sanitaires chinoises ordonnent la fermeture et l’assainissement du marché aux fruits de mer de Huanan à Wuhan (en fait la fermeture de plusieurs marchés annexes également). Dans ce marché de gros qui couvre toute la Chine méridionale, on y vendait, outre des fruits de mer, du gibier vivant tel que la fameuse civette palmiste qui avait fini par être identifiée comme l’hôte intermédiaire de la pandémie de SARS-Cov qui avait démarré à Canton en octobre 2002. Des conditions très similaires existaient concernant l’entreposage en grande promiscuité d’animaux sauvages comme des rats, des louveteaux, des blaireaux, des civettes palmistes et des pangolins, capturés ou issus en fait d’élevages clandestins. Tous ces animaux étaient forcément non contrôlés puisque l’objet de ventes en principe illégales. Selon les informations pas très fiables, rapportées et répétées dans les médias, on pouvait y trouver aussi, entre autres, des chiens, des serpents et même des chauves-souris. La présence d’une telle ménagerie au sein d’une mégapole de 11 millions d’habitants, officiellement illégale, mais officieusement acceptée par les autorités chinoises au titre de la tradition culturelle, est forcément un bouillon de culture naturel favorisant l’émergence de zoonoses comme celle du SARS-Cov de 2002-2003.
Du 1er au 12 janvier 2020, 585 prélèvements ont été analysés à l’Institut des maladies virales du Centre chinois de contrôle et de prévention des maladies (source : French.xinhuanet.com | Publié le 2020-01-27 ; Xinhua | 28.01.2020)
Désinformation de l’OMS probablement manipulée par la Chine
Malgré l’évidence de l’éminence avérée d’une épidémie à transmission interhumaine de la même nature que celle de SARS-COv de 2002-2003, le 12 janvier 2020, l’OMS, se fiant aux affirmations du gouvernement chinois, rapportera que : « à ce stade, […] il n’y a pas de preuve évidente de transmission interhumaine ». (sources : Commission municipale sanitaire de Wuhan · South China Morning Post · China Daily). Il s’agit là très vraisemblablement d’une première grosse intox, probablement involontaire de l’OMS manipulée par la Chine par l’intermédiaire de son président, le Dr Tedros Adhanom Ghebreyesus, élu Directeur général de l’OMS pour un mandat de 5 ans, en mai 2017. Cela prouve en tout cas un certain degré d’incompétence. En effet, l’hypothèse de base devant un virus qui s’apparente à celui de 2002-2003 est nécessairement de considérer, en premier lieu, la possibilité de contamination interhumaine et non pas l’inverse qui fait perdre un temps précieux dans le combat à mener.
À ce stade de l’épidémie le gouvernement chinois, qui espérait probablement que l’épidémie serait contrôlée facilement à Wuhan, avait tout intérêt à ne pas affoler le monde en attirant l’attention sur la possibilité bien réelle d’épidémie à répétition de type SARS-Cov provenant de son sol.
Il faut attendre le 20 janvier pour que la Chine annonce officiellement que le virus est transmissible et que Wuhan soit placée en confinement.
Il y a bien eu dissimulation de la contagiosité de la maladie respiratoire
En réalité, une personne clé au moins savait déjà bien avant le 20 janvier que le virus était transmissible d’homme à homme. Il s’agit de Huang Chao Lin, directeur adjoint de l’hôpital Jinyintan, premier hôpital désigné pour prendre en charge une pneumonie d’origine inconnue à Wuhan. Dans un article, publié dans Le Lancet, où il décrit les 41 cas de pneumonie atypique, extrêmement graves, recensés dans son hôpital, entre le 1er décembre et le 2 janvier, il comptabilise les cas exposés directement (27) ou non (14) au marché aux fruits de mer de Huanan.

Voici ce qu’il écrit : « Le premier cas fatal, qui avait eu une exposition continue au marché aux fruit de mer, avait été hospitalisé parce qu’il avait développé 7 jours de fièvre, toux, et trouble respiratoire (dyspnée). 5 jours après le début de sa maladie, sa femme de 53 ans qui n’avait pas été exposée au marché aux fruits de mer a développé une pneumonie et a été placée en chambre d’isolation. »
La seule conclusion vraisemblable que l’on pouvait tirer de ces données était que le syndrome respiratoire était contagieux visiblement.
Il décrit également la batterie de précautions utilisées pour se protéger des aérosols potentiellement générés par la maladie expectorés par les patients. Il est donc indéniable qu’avant le 1er janvier, dès l’alerte du 31 décembre, les autorités étaient au fait du caractère potentiellement très contagieux de la pneumonie. Il y a donc eu tromperie volontaire au niveau de l’OMS, ou par l’intermédiaire des autorités chinoises, qui informe le monde du caractère contagieux de l’épidémie seulement le 20 janvier, soit 3 semaines plus tard !
Localisation de l’épicentre au marché aux fruits de mer, une deuxième contre-vérité
Il est peu probable que l’unique épicentre de l’épidémie soit le marché aux fruits de mer de Wuhan. Le graphique des cas de pneumonies atypiques sévères, enregistrées au mois de décembre à Wuhan, montre clairement que le premier patient, dont les symptômes avaient débuté le 1er décembre, n’avait pas été exposé au marché aux fruits de mer de Huanan et n’avait pas également été en contact, selon l’enquête épidémiologique du Professeur Chaolin Huang, avec aucun des patients suivants. De même qu’il faut attendre 10 jours de plus pour avoir le début de symptômes du premier patient ayant été en contact avec le marché aux fruits de mer ; ce même jour correspond à 2 autres patients n’ayant eu aucun contact avec le marché aux fruits de mer.
Le premier cas connu aujourd’hui, après enquête rétrospective, remonterait au 17 novembre, une personne de 55 ans de la région de Hubei, n’a été diagnostiqué que rétroactivement cinq mois plus tard. Entre un et cinq nouveaux cas seront comptabilisés chacun des jours suivants, tous passés inaperçus (sources : Le Monde · Weixin · South China Morning Post). Malheureusement, on ne peut donc rien savoir des lieux que ces quelque 20 ou 30 patients ont fréquentés.
Mais à l’examen du tableau dressé à partir du 1er décembre, on se rend très bien compte que le début des symptômes des cas reliés au marché se concentre en fait entre le 15 décembre et le 27 décembre, avec 3 patients sur 4 n’ayant eu aucun contact avec le marché de Huanan dans la période des 2 semaines précédentes. Cela indique qu’il est très possible au moment où l’information est transmise aux autorités que le marché aux fruits de mer ne soit ni l’unique ni le premier épicentre de l’épidémie de Wuhan.
Pourtant, le 22 janvier, Gao Fu, directeur du Centre national de contrôle et des maladies déclare que l’épicentre du nouveau coronavirus à Wuhan est lié à la vente d’animaux sauvages sur le marché de fruits de mer à Wuhan en excluant la possibilité d’un foyer antérieur différent (source : transcription de la conférence de presse du Bureau d’information du Conseil d’État du 22 janvier 2020).
Par la suite, il a été montré que :
- Une étude rétrospective, parue dans le New England Journal of Medecine (NEJM), sur les 425 premiers cas de pneumonie de cause inconnue, confirmés rétrospectivement, dont les symptômes avaient débuté avant le 22 janvier, 45 % (26 pts) de ceux dont les symptômes avaient débuté avant le 1er janvier (soit 47 patients) n’avaient pas été exposés aux marchés des fruits de mer de Wuhan. L’enquête épidémiologique a été menée sur les patients ayant survécu et les proches des patients pour déterminer toutes leurs activités, leurs déplacements et leurs contacts potentiels avec des animaux sauvages pendant une période de 2 semaines précédant le début des symptômes.
- En fait, sur les 47 patients ayant présenté les premiers symptômes avant le 1er janvier, 30 (64 %) avait eu un contact avec un marché aux fruits de mer, soit le principal de Huanan (26 patients), soit un autre marché annexe (4 patients), 14 patients avaient été en contact avec un autre patient et 12 n’avaient eu aucun contact, ni avec un des marchés aux fruits de mer ni avec aucun des patients atteints du syndrome respiratoire.
La première déduction que l’on peut faire des chiffres présentés est qu’en fait 5 patients n’avaient pas été en contact direct avec un marché aux fruits de mer, mais en contact avec une personne présentant le syndrome respiratoire.
Cela confirme bien qu’au moment de l’alerte faite le 31 décembre (ou même avant) les autorités chinoises connaissaient certainement le caractère contagieux de ces pneumonies d’origine inconnue dont le système de surveillance avait été justement mis en place suite à l’épidémie de SARS de 2002-2003.
La deuxième déduction est que 12 patients présentant des symptômes respiratoires aigus avant le 1er janvier avaient donc été infectés dans une période de temps comprise entre 4 et 14 jours précédents, c’est-à-dire pour certains avant le 15 décembre, sans n’avoir jamais été en contact avec une personne présentant le syndrome respiratoire ni avoir été exposés à un des marchés aux fruits de mer de Wuhan.
On sait par ailleurs (voir plus haut) qu’il y a eu autour d’une vingtaine de cas (au plus 30) de pneumonies atypiques non détectés entre le 17 novembre et le 1er décembre sur lesquels il n’y a aucune information sur leur exposition à un marché aux fruits de mer. Il aurait fallu inclure dans l’étude rétrospective une enquête épidémiologique sur ceux de ces patients qui ont survécu ou sur leurs proches pour connaître leur exposition aux marchés aux fruits de mer ou à des personnes ayant été atteintes du syndrome respiratoire. Cette enquête n’a pas été faite alors que c’était, et c’est encore aujourd’hui parfaitement réalisable ! Connaissant la maîtrise et la rigueur scientifique des Chinois, on est forcé de conclure qu’il n’est pas dans l’intérêt des autorités de déterminer si les tout premiers cas de Covid-19 ont été exposés ou non aux marchés aux fruits de mer de la ville, ou ont été en contact avec une autre personne malade ou ayant ultérieurement développé la maladie.
Tout indique donc qu’on ne peut absolument pas exclure la possibilité d’un tout premier foyer initial (épicentre zéro réel) de l’épidémique à Wuhan autre que le marché aux fruits de mer de Huanan ou un des marchés annexes.
Destruction des preuves saisies sur le marché de Huanan
En réalité on ne sait pas vraiment officiellement le niveau de preuves saisies dans ce marché lors de sa fermeture puisque les autorités chinoises n’ont toujours pas rendu public le résultat de leurs analyses et ont détruit tous les échantillons prélevés lors de sa fermeture.
Quelle est la liste des espèces animales saisies et analysées ? Lesquelles étaient porteuses de coronavirus ? Quel degré d’identité génomique était partagé avec le SARS-Cov2 identifié à partir des patients ? Autant de mystères qui prouvent que la Chine retient certaines informations cruciales sur l’épicentre supposé d’une pandémie qui a ravagé la planète.
Quelles conclusions en tirer ?
La Chine contrôle parfaitement l’information, ce qui en fait est synonyme de désinformation.
Un exemple frappant de désinformation, parfaitement maîtrisée, mais éhontée, a été donné au monde lorsque le porte-parole du ministère chinois des Affaires étrangères, Zhao Lijian inversait les rôles en twittant le 12 mars : « Ce pourrait être l’armée américaine qui a apporté l’épidémie à Wuhan. Soyez transparents. Les États-Unis nous doivent une explication. » C’est une affirmation sans fondements, qui relève purement et simplement de la fake news, si ce n’est de la farce. ll n’est pas rare que même les athlètes de haut niveau soient malades en présentant des symptômes grippaux. En psychologie cela s’appelle inverser les rôles. En fait, c’est la Chine qui doit une explication au reste du monde, mais nos gouvernants sont trop lâches pour la demander officiellement.
Depuis 2003, tous les experts infectiologues du monde ont leurs yeux braqués sur la Chine en raison des marchés aux animaux sauvages, qu’elle refuse de fermer, ainsi que du vaste programme d’étude des coronavirus respiratoires véhiculés par les chauves-souris que l’armée chinoise, en parallèle des chercheurs universitaires, a commencé au début des années 2000.
La Chine a sans nul doute manipulé l’OMS en ce qui concerne la contagiosité de la maladie, retardant de 3 semaines une alerte qui aurait pu changer la donne mondiale à l’instar de l’épidémie de SARS de 2003 qui avait pu être jugulée à temps.
Elle dissimule aussi des informations concernant la localisation certaine du tout premier foyer de l’épidémie en évitant d’enquêter sur les cas hospitalisés qui étaient passés inaperçus, entre le 17 novembre et le 1er décembre, mais sur lesquels une enquête épidémiologique peut encore être conduite très facilement puisqu’ils ne sont qu’une trentaine au maximum, identifiés rétrospectivement.
L’étude des cas présentant leurs premiers symptômes entre le 1er décembre et le 31 décembre montre que le marché de Huanan n’est pas le seul foyer puisqu’il est prouvé que des marchés annexes sont également impliqués. C’est en contradiction avec la déclaration officielle du 20 janvier qui tend à ne parler que d’un seul marché aux fruits de mer, sous-entendant le principal, c’est-à-dire celui de Huanan, comme épicentre de l’épidémie.
Dans cette période, 12 cas sur 47 ne sont pas attribuables à un contact direct ni avec un marché aux fruits de mer ni avec une autre personne ayant développé la maladie. Cela indique la possibilité d’un autre foyer non encore identifié ou, forcément, de toute évidence la contagiosité interhumaine de la maladie en s’en tenant à l’hypothèse des marchés aux fruits de mer comme seul foyer initial de l’épidémie. Contagiosité prouvée dans cette période par le syndrome respiratoire aigu développé par la femme d’un homme lui-même décédé de la maladie alors qu’elle n’avait eu aucun contact avec un des marchés aux fruits de mer. On voit donc bien toute l’incohérence du discours des autorités chinoises et par conséquent celle de l’OMS.
D’autre part, la non-communication des résultats des tests biologiques effectués sur les animaux saisis sur le marché de Huanan et la destruction des échantillons prélevés démontrent la volonté de rétention d’informations cruciales. Ces informations pourraient éclairer les scientifiques du monde entier sur les causes de la pandémie et son mode de propagation.
De ce fait, la Chine entretient un flou artistique qui la met à l’abri des tracas que l’épicentre réel soit ou non le marché de Huanan.
Si la thèse du marché aux fruits de mer de Huanan comme épicentre principal tient, alors la Chine a de toute façon une responsabilité incontournable dans la genèse de l’épidémie de Wuhan, en plus de sa responsabilité dans sa transformation en pandémie mondiale incontrôlable. La Chine refuse depuis 2003 de fermer ses marchés aux animaux sauvages, souvent situés sur les mêmes sites que les marchés aux fruits de mer et donc sources potentielles d’épidémies mortelles de coronavirus. Il est prouvé que l’épidémie de 2002-2003 avait débuté dans le marché aux fruits de mer de Canton par l’intermédiaire de la civette palmiste, un gibier d’élevage.
Le gouvernement cache absolument que la Chine a failli dans sa responsabilité envers le Monde d’avoir un système de détection et d’alerte approprié. Le 4 mars 2019, Gao Fu, le directeur général du Centre chinois de contrôle et de prévention des maladies, au cours d’une réunion organisée à Pékin, la veille de l’ouverture des deux sessions du parlement, avait fait l’étonnante déclaration :
« Il y aura à l’avenir d’autres virus comparables au SRAS de 2003, mais il n’y aura plus d’épidémie comparable »
(source : Le Monde).
Chapitre 2
Partie 1 — La face cachée du laboratoire P4 de Wuhan
Les virus à gain-de-fonction, le paradigme caché de la recherche virologique récente
Dans le chapitre 1 de l’histoire du Covid-19, nous avons été sans concession pour dénoncer la responsabilité morale et politique de la Chine qui a pratiqué à outrance la désinformation d’État. Avec la complicité de l’OMS, elle a retardé de 3 bonnes semaines l’alerte au monde de la ré-émergence du syndrome respiratoire aigu sévère (SRAS) dû à un nouveau virus très similaire au SARS-Cov, extrêmement contagieux et responsable de l’épidémie mortelle de 2003. Nous avons expliqué que non seulement la Chine, mais aussi les gouvernants occidentaux, en particulier de la France et de l’Union Européenne, jouaient sur les mots en essayant de faire croire à l’arrivée soudaine d’une épidémie d’un genre nouveau, qui prendrait les populations de la terre par surprise, comme aux âges reculés de l’humanité, où les épidémies étaient perçues comme des fléaux imprévisibles qui s’abattaient aveuglément sur les populations.
La réalité est que les épidémies mortelles à caractère pandémique, comme celle que nous vivons à l’heure actuelle, sont anticipées et théorisées depuis le début des années 2000 par des consortiums oligarchiques financiers et par des états surpuissants tels que les USA et la Chine, mais également certainement la Russie plus discrète dans ce domaine. En raison de sa science et sa technologie de premier plan, nous verrons que la France s’est retrouvée également partie prenante dans ce domaine, en jouant un rôle de tout premier plan que nous décrivons dans la partie 2 de ce chapitre.
Ces trois très grandes nations dominantes ont développé à la fois des programmes militaires secrets, mais aussi des outils de surveillance tels que le National Scientific Advisory Board for Biosecurity (NSABB) aux USA, placé sous l’égide de l’autorité de santé civile américaine, le National Institute of Health (NIH).
Les états qui possèdent l’arme atomique envisagent la possibilité que dans un futur relativement proche des conflits mondiaux, mais surtout des attaques terroristes, puissent être perpétrés par l’intermédiaire de virus manipulés dont la virulence et la pathogénie auront été artificiellement renforcées par des modifications génétiques appelées gain de fonction (en anglais gain-of-function ou GOF).
Évidemment, moralement l’utilisation d’une telle arme ne peut pas être frontale. Elle pourrait justifier une réponse nucléaire en cas de menace où les intérêts vitaux ultimes d’un pays seraient menacés par un virus exterminateur. Mais, vice-versa, elle pourrait être une réponse militaire de dissuasion en cas de défaite militaire imminente ou même d’attaque du même ordre.
Ainsi, le Japon impérial aurait bien tenté de lancer désespérément en 1945 des milliers de ballons-bombes dont les nacelles devaient emporter initialement une charge bactériologique (un virus capable de tuer le bétail développé par le laboratoire secret Noborito ou bien l’anthrax où tout autre pathogène que le Japon avait déjà expérimenté et utilisé militairement auparavant) à destination des USA. La charge bactériologique a été ensuite remplacée par une bombe de 30 kg par crainte également de représailles bactériologiques, selon les historiens japonais. Le laboratoire militaire secret Noborito près de Tokyo, qui avait réalisé ce projet, est maintenant devenu un musée pour rappeler les concepts extrêmes auxquels les guerres conduisent. Les ballons devaient traverser l’Océan pacifique, poussés par les vents, et atteindre en 3 jours les côtes américaines. Quelques-uns y sont parvenus, semble-t-il. Cette attaque, réalisée avec des moyens assez réduits, mais non sans sophistication, a démontré que le concept avait été poussé jusqu’à réalisation.
De tout temps les armées ont su utiliser les lumières des ingénieurs et savants contemporains, de la renaissance à l’époque moderne. Léonard de Vinci, comme tous les savants et ingénieurs de son époque, essayait, entre autres, de gagner de l’argent en proposant des machines de guerre, toutes plus terrifiantes les unes que les autres, dont certaines étaient réalisables avec les techniques de son temps. Certaines planches de ses dessins faisaient état de machines volantes ou de cloches submersibles non encore réalisables. Cela faisait partie de la lutte pour la suprématie intellectuelle qui donnait une visibilité de prestige auprès du Prince qui pouvait faire commande de ces armes.
De nos jours, rien n’a changé, le projet Manhattan d’élaboration de la première bombe atomique a été réalisé par les plus grands physiciens de l’époque, comme Enrico Fermi et Robert Oppenheimer qui se sont retrouvés sur la vaste zone militaire secrète de Los Alamos, dans le désert du Nouveau-Mexique, pour y diriger la fabrication de la première bombe de l’histoire.
Les militaires ont conscience qu’ils n’ont aucun génie ni aucune capacité dans les domaines scientifiques. Ils se contentent bien plus efficacement de confier aux savants universitaires et aux institutions civiles la tâche de développer les technologies d’avant-garde pouvant permettre de développer des armes nouvelles, quitte à les surveiller discrètement. Indirectement, ils suscitent chez quelques universitaires très imaginatifs un intérêt certain pour mettre au point, quasiment en toute liberté, les techniques donnant la possibilité de créer in fine des armes terrifiantes. Cela se fait au prétexte que l’on ne peut arrêter le progrès ou bien, comme nous le verrons, au prétexte de la lutte contre les pandémies.
Contrairement à ce que le grand public croit, le sens moral et la conscience de la portée possible des actes de recherche ne sont pas l’apanage consubstantiel de la connaissance scientifique. Sans même parler de l’attrait du prestige et de la gloire des découvertes dans la psyché des chercheurs, le déni des scientifiques passe souvent par l’affirmation que toute découverte peut être utilisée indistinctement à des fins utiles ou nuisibles. Et qu’ainsi leur responsabilité ne peut être réellement engagée puisque leur statut de chercheur les pare automatiquement des meilleures intentions. Les exemples concrets que nous allons développer montrent l’actualité brûlante du célèbre proverbe de Rabelais : « La sagesse ne peut pas entrer dans un esprit méchant, et science sans conscience n’est que ruine de l’âme ».
Les états et les militaires pensent de leur côté qu’il vaut mieux maîtriser les avancées technologiques en premier quitte à en interdire ensuite l’accès aux autres nations. La prolifération de l’arme nucléaire en est l’exemple flagrant qu’il est inutile de commenter tant les moyens extrêmement lourds, que le développement de cette arme nécessite, rendent sa dissémination contrôlable.
En ce qui concerne les virus manipulés, le contrôle est plus difficile. Les trois grands états dominant le monde savent que cette technologie n’est pas encore d’accès généralisé à l’ensemble des pays, car elle nécessite beaucoup de moyens intellectuels et techniques très diversifiés. Il faut de l’expertise, du savoir-faire, des moyens financiers, des laboratoires spécialisés (en principe de haute sécurité, mais pas nécessairement) et une armée de techniciens et de laborantins très bien formés, travaillant sous la direction de chercheurs compétents. Nous verrons dans la partie 2. Comment la France a fourni à la Chine tout cela dans le cadre d’une coopération civile et universitaire.
Les USA, la Chine et la Russie savent qu’aucun d’entre eux n’a réellement intérêt à un conflit ouvert de ce genre. De ce fait, la crainte principale du monde occidental est limitée depuis l’épidémie mortelle de SARS de 2003 à ce qu’un pathogène à potentiel pandémique (PPP) puisse s’échapper par accident d’un laboratoire. Un témoignage de la réalité fondée de cette crainte apparaît dans une note du NSABB datant de 2013, une année charnière où certaines institutions commencent à comprendre le danger. Elle stipule, pour ceux qui auraient encore la naïveté de croire le contraire, qu’il n’existe aucune régulation ni surveillance réelle en matière de recherche GOF et qu’un virus échappé de laboratoire peut créer une pandémie avec des millions de morts. Cette note mentionne l’existence d’une liste documentée de virus mortels échappés accidentellement de laboratoires par le passé et ayant causé un nombre de décès conséquent.

Virus échappés de laboratoire par le passé
Nous référons le lecteur au remarquable, mais quelque peu inquiétant article : « Une brève et terrifiante histoire de virus s’échappant des laboratoires » publié le 16 avril 2014 (source SlateFr). Le contenu de cet article documenté et sourcé est hautement édifiant et ne peut que très difficilement être mis en doute. Il confirme l’implication des trois mêmes grandes nations dans la ré-émergence de virus pathogène du passé.
Par exemple, il explique en détail comment la grippe porcine, un virus de type H1N1 apparenté à celui de la grippe espagnole de 1918, s’est selon toute vraisemblance échappé d’un laboratoire militaire à Fort Dix dans le New Jersey en 1976. Puis, de nouveau, un virus de type H1N1 réapparaît aussi comme par hasard en Russie et en Chine en 1977. Les analyses virologiques et génétiques de l’époque permettaient de suspecter une fuite de laboratoire d’un virus datant des années 1940 à 1950. Cela s’est trouvé confirmé plus tard par des études génomiques. SlateFr écrit : « En 2010, cette confirmation devient un fait scientifique : “Le cas le plus célèbre d’une souche virale échappée d’un laboratoire concerne la réémergence de la grippe A H1N1, observée pour la première fois en Chine en mai 1977, et quelque temps après en Russie”, affirment des chercheurs. ».
Nous ajoutons que ce fait est démontré dans un article publié dans PlosOne une revue scientifique de tout premier plan. Il est démontré qu’il manquait des décennies d’évolution dans les échantillons prélevés en 1977 ce qui prouvait qu’il s’agissait de la réémergence d’un virus stocké en laboratoire ! En effet, un virus stocké dans des éprouvettes en laboratoire est dormant, il ne se réplique pas et donc ne mute pas.
En ce qui concerne l’incident de Fort Dix, qui a causé la mort d’un soldat et la contamination de deux autres, il n’y a jamais eu de déclaration officielle du gouvernement américain au sujet d’un accident de laboratoire. Une chose est sûre, la souche de virus identifiée, après prélèvements sur le soldat mort et les deux autres contaminés à Fort Dix, a suffisamment fait peur aux autorités américaines pour que l’OMS soit informée et que 48 millions d’Américains, soit 22 % de la population, soient vaccinés, avant que le programme ne soit arrêté. En effet, aucun autre cas n’avait été enregistré entre temps, confirmant implicitement d’ailleurs la cause probable d’un accident de laboratoire.
Cette campagne a rapporté évidemment beaucoup d’argent aux sociétés pharmaceutiques Sharp & Dohme (Merck), Merrell, Wyeth, et Parke-Davis qui avaient refusé de vendre à prix coûtant le vaccin au gouvernement américain. Après coup, une fois la panique passée, la vaccination était apparue très discutable, car, comme toute vaccination, elle a eu un coût humain : les autorités de santé ont enregistré 532 cas de syndrome de Guillain-Barré imputés à la vaccination et 25 morts directement suite aux injections.
Ce qu’il faut comprendre et comme il est très bien expliqué dans l’article paru dans SlateFr : « la souche de grippe humaine H1N1 est apparue avec la pandémie mondiale de 1918 pour, lentement, accumuler ensuite de légères modifications génétiques, et ce jusqu’en 1957, où elle fut considérée comme disparue après l’émergence du virus pandémique H2N2. » La proportion de mutations entre deux virus dérivant l’un de l’autre permet d’établir une horloge moléculaire qui, une fois calibrée, donne une assez bonne estimation de la période de temps qui a pu s’écouler entre deux émergences épidémiques d’un même virus. C’est donc un outil d’investigation qui permet de vérifier si la résurgence d’un virus ancien est naturelle ou provient d’un laboratoire.
De nombreux laboratoires conservent des souches de virus H1N1 et quelques-uns d’entre eux, des souches initiales de la pandémie de 1918-1919, obtenues en déterrant des morts enterrés dans le permafrost en Alaska. En 2005, une équipe acheva ce projet en séquençant le génome complet du virus (SlateFr). Ce virus se trouve à présent à l’abri dans des laboratoires P4 !
Il faut noter que ces cas historiques ne concernent évidemment que des virus naturels, non manipulés génétiquement, mais laisse entrevoir la dangerosité encore plus évidente dans un futur proche d’accidents de virus à gain-de-fonction, dont la contagiosité et la pathologie auront été artificiellement augmentées.
Comme nous allons le voir dans la section suivante, dédiées à nos apprentis sorciers modernes, la formule de l’article de SlateFr résume parfaitement la situation à venir : « L’ironie de la chose, c’est que ces établissements [les institutions de recherches impliquées, NDLR] travaillaient sur ces pathogènes dans le but de prévenir les épidémies qu’ils allaient eux-mêmes provoquer. Leurs conséquences tragiques ont donc souvent été qualifiées de « prophéties auto-réalisatrices ».
Les apprentis sorciers du 21e siècle
En parallèle aux tâtonnements intellectuels des militaires et des états, des apprentis sorciers de tout poil sévissent dans les laboratoires universitaires de recherche en virologie et microbiologie en y faisant de nombreux émules. Pour le prestige de la découverte scientifique, pour une carrière universitaire du moins, ou pour l’argent oligarchique, ils sont prêts à procéder à toutes les manipulations génétiques possibles de virus afin d’augmenter certaines de leurs fonctions comme la contagiosité ou la pathogénie dans le cadre général de programme de recherche visant officiellement à prévenir les pandémies. Les autorités civiles de santé américaine (NIH) se sont apparemment, mais assez faussement, émues de cette situation en 2013 à la suite des révélations sur la recherche gain-de-fonction du Néerlandais Ron Fouchier. (voir le reportage du site plandemicseries).
L’émotion est grande dans certains milieux informés et le NIH décide de suspendre le financement des recherches à gain-de-fonction sur les virus en 2013, recherches qui ne sont pas toutes, loin de là, effectuées dans des laboratoires P4 de plus haute sécurité, mais dans des laboratoires P2 et P3 dans les universités sont équipées, à l’instar de la recherche de Ron Fouchier à l’Université Erasmus de Rotterdam (dont le laboratoire P3 à été transformé en P3+ à atmosphère à pression négative sans pour autant atteindre le niveau P4) et de Yoshihiro Kawaoka aux USA. Les laboratoires P2 et P3 présentent des conditions de sécurité et de confinement bien moindre que les P4, en particulier ils ne sont pas mis en régime atmosphérique permanent de pression négative et ne sont pas équipés de double sas d’entrée, avec douche obligatoire, qui empêchent en principe toute échappée de pathogène vers l’extérieur par voie aérienne.
Cependant, en 2013 le NIH n’avait aucune intention de stopper réellement les recherches GOF sur les virus tels que le H5N1 ou le H7N9 et, duplicité oblige, les programmes de recherche auxquels des bourses du NIH avaient déjà été allouées pouvaient continuer. Ce qui voulait dire en termes pratiques, puisque les programmes de recherche US sont subventionnés sur une durée de 2 à 3 ans, que les recherches gain-of-function pouvaient continuer tranquillement sur les virus les plus pathogènes, comme celui du SARS de 2003, encore pendant 2 ans minimum. Et c’est ce qui s’est passé. Ensuite à partir de 2015, en raison de pression provenant probablement de chercheurs et d’hommes publics conscients du problème, le NIH a choisi, comme pis-aller pourrait-on dire, d’externaliser les recherches sur les virus gain-of-function par l’intermédiaire d’un « portage » vers une ONG internationale, EcoHealth Alliance Inc. basée à NewYork, servant de couverture au financement d’instituts universitaires situés à Singapour et en Chine, dont l’Institut de Virologie de Wuhan et son laboratoire P4 fourni par la France qui venait de rentrer en service officieusement.
En 2013, malgré la pression morale, le NIH ne tenait visiblement pas à abandonner les recherches sur les virus GOF. Mais aujourd’hui, à l’heure de la pandémie de Covid-19, le NIH a confirmé publiquement au magazine USA Today que la bourse de recherche allouée à EcoHealth Alliance Inc. a été stoppée définitivement. Elle s’élevait à 3.4 millions de dollars sur 6 ans redistribués par EcoHealth Alliance Inc., le principal récipiendaire, aux sous-récipiendaires affiliés : Wuhan Institute of Virology (Wuhan), East China Normal University (Shanghai), the Institute of Pathogen Biology (Pekin), et Duke-NUS Medical School (Singapour).
Les chercheurs universitaires néerlandais, Ron Fouchier, et américain Yoshihiro Kawaoka, créent un virus synthétique de grippe aviaire potentiellement pandémique. Chacun de leur côté, le Dr Ron Fouchier, néerlandais, et le Dr Yoshihiro Kawaoka, américain, ont publié en 2012, le résultat de leur recherche sur la création d’un virus de grippe aviaire H5N1 hautement pathogène, génétiquement modifié par mutations sélectives et transmissible entre mammifères (le furet) par l’intermédiaire de gouttelettes respiratoires. Ces recherches ont soulevé un certain émoi et une controverse dans la communauté scientifique travaillant sur les virus de grippe aviaire dont une partie a volontairement suspendu certaines études GOF sur le virus H5N1.
Les virus obtenus par ces deux chercheurs ont été fabriqués à partir de la souche initiale hautement pathogène du H5N1 (grippe A), un virus respiratoire, transmis à l’homme par l’intermédiaire de contacts rapprochés avec des élevages de volaille, eux-mêmes contaminés par des oiseaux sauvages migrateurs. Le H5N1 a un taux de létalité affolant de 60 %, mais fort heureusement n’est pas transmissible entre humains… enfin, pas encore tout à fait puisque Fouchier et Kawaoka ont décidé de lui conférer le caractère de transmissibilité entre mammifères seulement entre furets et pas entre hommes (éthique oblige). Ils ont choisi le furet, car c’est l’animal de laboratoire dont le système respiratoire est le plus proche de l’homme. Pour l’instant ces virus sont créés et sont stockés dans des laboratoires P2/P3, mais combien de temps encore avant qu’ils ne soient relâchés dans la nature par accident et par la suite, directement ou par adaptations successives, finissent par franchir la barrière des espèces vers l’homme ?
À la suite de ces publications, 22 virologistes ont notifié la communauté de recherche de leur intérêt pour la création de souches transmissibles entre humains du virus mortel H7N9 de la grippe asiatique apparu au printemps 2013 en Chine, tuant 43 des 130 personnes infectées.
En 2013, Fouchier à Rotterdam et Kawaoka à l’Université du Wisconsin-Madison remettent le couvert en proposant une étude GOF de manipulation génétique sur le virus H7N9 pour le rendre plus pathogène, plus résistant aux antiviraux et transmissible entre mammifères.
“Avec ces expériences, ils espèrent « trouver ce qui rend cet agent pathogène potentiellement mortel pour l’homme et les moyens d’arrêter sa possible propagation »” (Le Figaro). La sempiternelle ritournelle de nos apprentis sorciers n’est pas sorcière : apporter des bienfaits à l’humanité.
Dans son rapport, Lynn C. Klotz, chercheur confirmé (Senior Fellow Scientist) au Centre de Contrôle de Prolifération des Armes (Center for Arms Control and Non-proliferation, USA) écrit : “Ces deux chercheurs ont marqué le début l’ère des entreprises de recherches visant à créer en laboratoires des pathogènes à potentiel pandémique (PPP)”. Elle recense pas moins de 35 publications, la plupart issues de recherches en Asie, décrivant la création de PPP ou d’expériences conduites autour de PPP. Lynn C. Klotz a par ailleurs publié un article scientifique décrivant les conséquences pour le monde d’un accident de laboratoire qui libérerait un virus pathogène à potentiel pandémique.
Le Docteur Shi Zheng Li et le Professeur Ralph S. Baric vont encore plus loin en 2015, en créant un virus COVID synthétique hautement pathogène
Dans un précédent mini-article publié le 8 juin 2020, FranceSoir informait le grand public qu’un virus COVID synthétique hautement pathogène pour l’être humain avait été créé dès 2015 par l’institut de virologie de Wuhan en collaboration avec l’Université de Chapel Hill (Caroline du Nord) aux USA. Les preuves sont indiscutables puisque la recherche a été publiée en décembre 2015, avec force détail, dans le très sérieux journal scientifique anglais Nature Medicine.
Dans cet article, Shi Zheng Li, directrice du laboratoire des pathogènes spéciaux du laboratoire de virologie de Wuhan et le Professeur Ralph Baric décrivent comment, suite à une découverte majeure faite par Shi Zheng Li dans son laboratoire de Wuhan, ils ont pu créer un coronavirus hybride entre le virus du SARS de 2003 et la protéine S d’un coronavirus de chauve-souris identifié au laboratoire de virologie de Wuhan. Ce virus infectait les cultures de cellules respiratoires humaines avec les mêmes niveaux mortels que ceux observés chez les patients atteints du SARS en 2003.
Cette recherche publiée est si dérangeante pour les médias dits « mainstream », contrôlés par l’oligarchie financière, qu’une véritable omerta existe en France autour de cette expérimentation GOF. Elle est devenue un sujet tabou que l’on ne peut aborder sans être immédiatement frappé d’excommunication. FranceSoir a eu d’ailleurs maille à partir avec NewsGuard, en se voyant décerner un label rouge par ce site internet américain qui traque les fake news et se targue de combattre le complotisme.
Comme nous allons le voir dans le chapitre suivant, Shi Zheng Li est en réalité à la fois la grande prêtresse et le cerveau dans la création de ce virus Covid synthétique. Son temple est le laboratoire P4 de Wuhan que la France lui a en quelque sorte offert, après lui avoir décerné un doctorat de microbiologie à l’Université de Montpellier au début des années 2000, à l’Université de Montpellier où elle était arrivée en 1998.
La France après avoir livré clé en main à la Chine un laboratoire P4 perd le contrôle de ce qui s’y passe et est flouée

Les tenants et aboutissants de ce qui, d’ores et déjà apparaît comme le point final d’une déplorable aventure d’alliance stratégique de l’INSERM et des laboratoires Mérieux avec la Chine dans le domaine de la fabrication de vaccins pour le contrôle des pandémies futures, seront bien difficiles à reconnaître par les autorités françaises qui, apparemment, ont fait preuve d’une certaine légèreté.
Ainsi, la France a tenté de développer avec la Chine, un projet de politique extérieure ultra-sensible de contrôle des épidémies émergentes, devenu légitimement une priorité depuis le SARS de 2003. Un résumé circonstancié, très détaillé, a été publié le 17 avril 2020 dans un article de France Inter relatant l’historique de la création de ce laboratoire P4. Ce projet exceptionnel avait été approuvé en 2003 sous la présidence de Jacques Chirac. Ce projet s’est appuyé sur l’expertise du laboratoire INSERM Jean-Mérieux à Lyon, considéré parmi les tout meilleurs du monde.
Conçu sur le modèle du laboratoire P4 de Lyon, sa construction a été mise en œuvre par des entreprises françaises de haute technologie, en 2008, après d’interminables discussions dues à des réticences, comme en témoigne le Pr Christian Bréchot, directeur de l’INSERM de l’époque. Son inauguration officielle, en grande pompe, en même temps que son accréditation a eu lieu le 23 février 2017 sous l’égide du Premier ministre de l’époque, Bernard Cazeneuve, accompagné de la ministre française des Affaires sociales et de la Santé Marisol Touraine, ainsi que d’Yves Lévy, président de l’INSERM depuis 2014.
Signalons cependant qu’il s’avère à présent que le laboratoire P4 de Wuhan n’est plus le seul dont dispose la Chine. L’annonce de la mise en service d’un laboratoire vétérinaire de très haute sécurité P4 à Harbin, dans la province de Sahaliyan Ula en Mandchourie, a été publiée le 8 août 2018 par une agence communication franco-chinoise. Selon un journaliste de Challenges, cette mise en service serait en contradiction avec les engagements pris par la Chine. Cette dernière assertion est difficilement vérifiable et très discutable, car la nature des accords, passés avec la Chine, qui ont présidé à la création du P4 de Wuhan n’est pas connue du public, ce qui pose problème dans notre démocratie.
En effet, la limitation de la capacité d’un pays dans un domaine technologique à portée sanitaire (voire militaire) nécessite la signature d’accords internationaux officiels (à moins d’un accord secret signé, dont la valeur serait également discutable). Il ne s’agissait probablement, que d’une promesse verbale, sans réelle valeur juridique, faite aux autorités françaises dont on peut se demander si elle n’ont pas fait preuve d’une certaine de naïveté de la tenir pour argent comptant. La Chine a bel et bien acquis ainsi une technologie et un savoir-faire qu’elle peut maintenant développer à sa guise. Notons au passage, que sur les 45 laboratoires P4 dans le monde, 9 ne se situent pas dans des pays occidentaux ou d’influence occidentale : 3 en Russie, 3 aux Indes, 1 en Biélorussie et donc 2 en Chine !
L’affirmation faite à FranceInfo par Thierry Breton, ancien ministre de l’Industrie, et aujourd’hui commissaire européen, au sujet de la gestion de la crise du coronavirus par la Chine : « Nous ne sommes pas naïfs avec la Chine et nous ne l’avons jamais été », s’apparente à un déni rhétorique ! On a envie de lui répondre que « gouverner, aurait été de prévoir » et l’on pourrait aisément étendre ce constat aux gouvernements successifs, qui ont présidé à cette tentative de coopération désastreuse avec la Chine.
La dotation de la Chine d’un laboratoire de haute sécurité P4 s’est accompagnée du transfert d’un savoir-faire à très forte valeur ajoutée, par l’intermédiaire de la formation de techniciens (formation effectuée d’ailleurs au laboratoire P4 conjoint de l’INSERM et Merieux à Lyon) et de personnels scientifiques très compétents, à l’instar de la très brillante docteur en microbiologie, Shi Zhen Li, elle-même formée à la faculté des sciences de Montpellier.
Mérieux et les gouvernements français voyaient certainement dans ce transfert de technologie à la Chine et la création d’une coopération de recherche, le moyen d’être en première ligne par rapport à la connaissance des virus hautement infectieux. En même temps, cela permettait à Mérieux d’avoir un accès direct au matériel viral, en vue de la mise au point de vaccins contre les pandémies mondiales redoutées. Les dizaines de milliards d’euros que peut rapporter la vaccination de milliards de personnes sur la terre au cours d’une pandémie valaient bien le risque que représentait ce projet ultra sensible.
La réalité c’est qu’une fois le laboratoire P4 mis en service, la Chine n’a pas tenu ses engagements et la cinquantaine de scientifiques et techniciens français qui devait travailler à cette coopération, selon les accords conclus, n’ont pas pu s’y rendre. Cela a fait dire à Alain Mérieux à Radio France à Pékin : « J’abandonne la coprésidence du P4 qui est un outil très chinois. Il leur appartient, même s’il a été développé avec l’assistance technique de la France ». Au final, la France a fait chou blanc, comme s’en excuse d’ailleurs Mérieux : « Il est impensable que la Chine n’ait pas un laboratoire de haute sécurité pour isoler des germes nouveaux dont beaucoup sont d’étiologie inconnue. »
La France n’a donc absolument rien reçu en retour, même pas la reconnaissance de la Chine, comme en témoigne le manque patent de transparence des autorités chinoises, et concomitamment la recommandation désastreuse de l’OMS de garder les frontières ouvertes, alors qu’un virus mortel se répandait sur la planète.
À présent, le laboratoire P4 de Wuhan est devenu une tour d’ivoire transformée en bunker, où absolument aucune personne étrangère n’a accès et d’où aucune information ne peut filtrer au dehors. Et dire qu’au départ, on croyait que seuls les virus ne devaient pouvoir s’en échapper ! Le régime de Pékin y exerce une censure absolue sur ce qui s’y est passé ou a pu s’y passer. Sans l’article publié en 2015 dans le journal Nature Médecine, la Chine serait en position de nier totalement d’avoir procédé à quelque manipulation de virus que ce soit.
Pour finir avec ce tableau désolant, nous rappellerons avec ironie l’article signé en 2017 par le directeur de laboratoire P4 conjoint « INSERM-Mérieux, paru dans Science et Santé à la rubrique « Stratégies » qui titrait : « Le laboratoire P4 de Wuhan, une réussite pour la coopération franco-chinoise » ! Il n’y a, semble-t-il, rien à ajouter, si ce n’est qu’en démocratie les politiques devraient normalement rendre des comptes sur les accords internationaux non tenus et leurs implications. Cela ne semble pas être le cas avec ce fiasco géopolitique et industriel français.
Chapitre 3
Partie 1 — La face cachée du laboratoire P4 de Wuhan
Credo de l’homme à l’image de dieu et la saga des apprentis sorciers
La pensée des états est dominée par le credo illusoire de la science toute puissante véhiculé par des conseillers scientifiques qui ressemblent plus à des éminences grises soumises à des influences obscures, qu’à d’éminentes personnalités publiques largement reconnues pour leur réalisation et leur mérite.
Le credo de l’homme tout puissant qui contrôle la nature
Qu’on le veuille ou non, le débat sur les recherches à gain-de-fonction ne peut être réduit à la seule rationalité comme le voudraient bien certains apôtres de l’idée d’un être humain affranchi des contraintes imposées par la nature, voire même la dominant à l’image de dieu, si tant est qu’il existe. Il s’agit là du prolongement de la vision d’un monde moderne, industrieux, tout puissant, introduite à la faveur de la réforme calviniste dont la doctrine était centrée sur un homme à l’image de dieu (imago Dei), mais dont il ne lui resterait que des vestiges. Cela s’entend avant tout sur le plan spirituel bien entendu. Bref, il ne s’agirait là que d’un débat théologique de plus de l’histoire de l’humanité, si la doctrine de Calvin n’avait pas autant marqué profondément le monde anglo-saxon, très pragmatique, jusque dans l’exploitation de concepts philosophico-religieux à des fins terrestres. N’oublions pas la place centrale qu’occupe la religion dans la vie et la mentalité américaine, les présidents américains prêtant encore serment sur la Bible à leur entrée en fonction…
Pour les Américains, la forme d’athéisme français, où l’on peut développer une morale dans l’humanisme en dehors toute religion, est inconcevable. En ce qui concerne, imago Dei, un théologien protestant moderne américain, Gregory Beale, s’est empressé de clarifier la situation pour ceux qui auraient eu des doutes sur la portée terrestre de ce concept de théologiens. « Mais pour Gregory Beale nous pouvons encore aller plus loin », rappelle le Bon Combat, un magazine religieux français, dans un article sur le sujet. En effet, on y lit que : « l’image de Dieu » n’est pas seulement liée à notre être, mais aussi à notre faire. » Et puis encore : « L’image de Dieu passe donc aussi par le fait de soumettre, dominer, et remplir la terre : 3 activités parallèles à l’activité de Dieu lors de la Création qui soumet le chaos, domine en faisant toute chose, et remplit la terre avec tout ce qui s’y trouve. »
Fort heureusement, cette vue du protestantisme dépassant le cadre théologique n’est pas forcément épousée par l’ensemble des populations anglo-saxonnes. En effet, de nombreuses voix de bon sens se sont élevées également aux USA, et continuent de s’élever, pour dénoncer ces recherches qui font peser un grave danger sur les populations de notre planète.
Cependant, on se rend donc compte de la portée morale et philosophique des recherches GOF sur les virus. Il est extrêmement tentant de fabriquer des virus actifs, encore plus actifs, c’est fascinant, c’est passionnant ! Car la fonction des virus c’est d’être pathogènes et virulents et donc le chercheur se sent tout puissant de pouvoir se substituer à la nature, être plus puissant qu’elle en perçant ses mystères biologiques les plus intimes. Malheureusement, in fine, malgré l’imago Dei d’inspiration protestante, c’est la nature et les événements qui auront certainement le dernier mot sur nos apprentis sorciers.
L’affirmation de certains d’entre eux que les recherches GOF permettront de combattre les pandémies du 21e siècle tant redoutées n’a quasiment jamais été contredite officiellement avant la pandémie actuelle. Pourtant, cette affirmation ne tient pas a priori lorsqu’elle est passée au crible de la simple raison.
La peur peut engendrer des catastrophes
Tout armurier comprend qu’il n’y a pas besoin de transformer une arme semi-automatique en arme automatique pour comprendre en quoi elle est mortelle. Elle est mortelle quand le canon pointe vers une personne, qu’une balle est engagée dans la culasse et que percuteur est activé par la détente. Si l’on retire chacun de ces éléments séparément, l’arme ne fonctionnera pas.
Un armurier exalté pourrait se sentir investi d’une mission particulière dans l’existence. Celle de protéger ses semblables des armes à feu potentielles qui pourraient avoir une puissance de feu décuplée avec un système de réarmement et de percussion entièrement automatisé. Pour prouver au reste du monde qu’il y a des raisons d’avoir peur, il décide, alors que rien ne l’y oblige, de fabriquer cette arme et de révéler au monde la façon exacte dont il a procédé, pour ensuite la ranger en lieu sûr ! Par ses actes a-t-il ainsi contribué à lutter contre l’existence d’une telle arme ou bien a-t-il participé à l’accélération de sa création et de sa dissémination ?
On voit que l’excès, comme le manque d’imagination, peut tuer de façon égale. L’armurier croit-il vraiment être le seul homme à pouvoir concevoir et réaliser une telle arme pour en protéger le monde au cas où il adviendrait que quelqu’un d’autre de mal intentionné la réalise ? Développer une telle réflexion de justification n’est-ce pas là le signe évident d’une pathologie mentale ? D’un ego surdimensionné ? Dans l’histoire de l’humanité, la révélation d’une découverte a toujours permis sa reproduction rapide par d’autres personnes ou groupes de recherche. Ce phénomène d’accélération de la dissémination est concomitant de sa révélation au monde.
En ce qui concerne les dangers que recèle la nature avec des virus en perpétuelle évolution, une analyse similaire de comportement peut s’appliquer à nos chercheurs scientifiques, apprentis sorciers. Le résultat de leurs actes résultant d’une peur mal maîtrisée des pandémies peut engendrer des pandémies. Les accidents de laboratoires évoqués dans le chapitre précédent (chapitre 2) le prouvent.
Les arguments de peur
Les mécanismes biologiques qui régissent l’activité des virus sont certes d’une nature bien plus complexe et difficile à établir que ceux d’une arme à feu. Cependant, le principe d’étude par perte de fonction, ou par l’intermédiaire d’autres approches de la biologie moléculaire, au lieu de gain de fonction, pourrait tout à fait être mis en œuvre comme cela est fait par l’immense majorité des équipes de recherche travaillant à la compréhension des virus. Cependant, ce principe de bon sens se heurte à la contre-argumentation spécieuse de Ron Fouchier et d’autres qui mettent en avant principalement que le temps presse.
Argument de peur de Ron Fouchier : le virus de la grippe A/H5N1 pourrait muter et devenir transmissible d’homme à homme
Ron Fouchier écrit avec mauvaise foi à propos du virus de la grippe A/H5N1 : « Savoir si ce virus peut acquérir la capacité de se transmettre par aérosols ou gouttelettes respiratoires entre mammifères, y compris les êtres humains, pour déclencher une pandémie future est une question clé pour s’y préparer. ». En conséquence de son affirmation, il a donc créé en laboratoire un tel virus pour prouver que c’est une réalité à laquelle il fallait se préparer.
Dans son article, il n’indique pourtant absolument pas en quoi la création d’un virus H5N1 hautement pathogène muté pour être transmissible entre furets, un animal dont le système respiratoire est très proche de celui de l’être humain, va permettre de mieux le combattre ? Silence radio total sur ce sujet pourtant hautement intéressant ! Pourquoi ?
En réalité, cela n’est pas dit, mais l’idée est d’essayer de développer des vaccins sur des animaux de laboratoire, sur les furets au départ, mais également sur d’autres animaux tels des singes, avec de forte chance, mais sans garantie absolue que la méthode vaccinale mise au point puisse être portée à l’homme en cas de nécessité.
Les mutations engendrées sont sur une région exposée et donc antigénique, ce qui veut dire qu’on pourrait potentiellement générer des anticorps qui vont pouvoir la reconnaître spécifiquement. Mais cela implique beaucoup de manipulation de virus et la contamination forcée de dizaines d’animaux de laboratoire, multipliant les chances de fuite du virus vers l’extérieur ou de contamination accidentelle d’un laborantin. Au final quelle importance s’il y a fuite du virus si le vaccin est prêt, pourrait-on dire non sans cynisme ! Une chose que le grand public ne sait pas, et que le gouvernement s’acharne à masquer, c’est qu’un vaccin n’est jamais fiable à 100 % et ne présente jamais son niveau de sécurité parfait. D’ailleurs : « Un haut dirigeant de la société pharmaceutique AstraZeneca confirme désormais à Reuters que le géant pharmaceutique britannique s’est vu accorder l’immunité de toute plainte légale, au cas où le vaccin d’AstraZeneca devait finalement montrer des effets secondaires nocifs. » (source : BusinessAM)
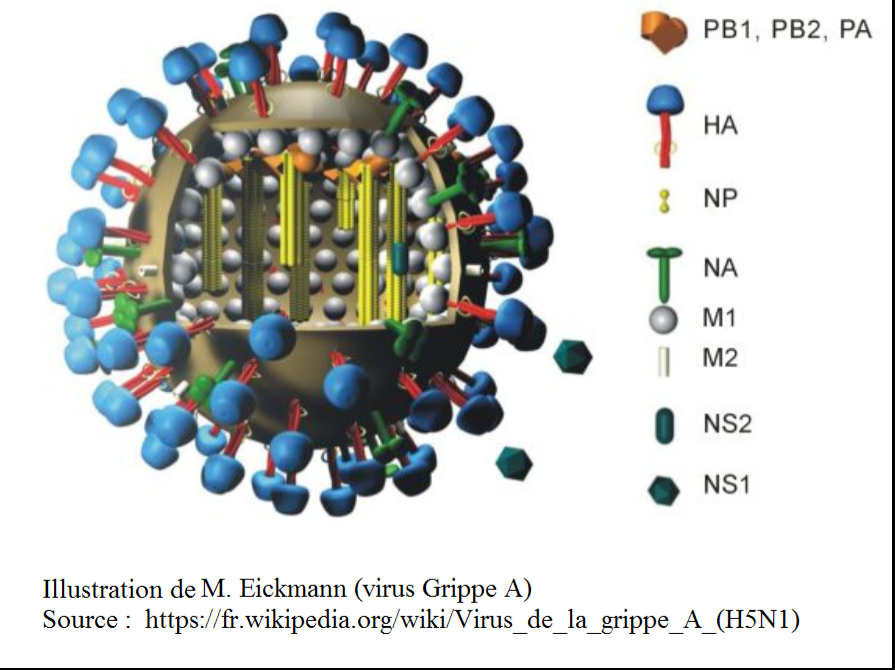
Par contre, le fait est qu’en créant ce virus pathogène et contagieux pour le furet, et parfaitement viable, il l’a quasiment créé également pour infecter l’homme, en plus d’indiquer la recette pour rendre les virus grippaux transmissibles par voie aérienne. Le furet est un intermédiaire particulièrement actif par ce qu’il peut être également infecté par des virus de grippe humains et les recombiner avec ses propres virus pour en faire des hybrides naturels qui seront capables d’infecter à la fois le furet et l’homme. Cela en fait un hôte intermédiaire de premier choix.
Cependant, l’argument de Fouchier est étrange puisqu’il recherche les mutations susceptibles de produire un virus pandémique, car, dit-il, des recombinaisons H5N1 aviaire et les virus de grippe A (influenza) humaine, ne sont pas transmissibles entre furets par voie aérienne et que, d’autre part, ne se produisent pas naturellement via d’autres intermédiaires animaux entre le H5N1 aviaire et les virus de grippe A (influenza) humains. Donc, en fait, Monsieur Fouchier, le danger de recombinaison naturelle dans un hôte intermédiaire proche de l’homme n’est donc pas si imminent que ça.
La seule voie de transformation hautement contagieuse du virus vers l’homme passerait donc forcément par des mutations naturelles qui lui permettraient d’activer les récepteurs de pénétration viraux de la grippe plus spécifiques à l’homme. Mais quelles sont les chances que cela arrive naturellement ? Fouchier ne donne pas la probabilité qui pourtant peut s’estimer (voir plus loin dans l’article).
Quelle est la fonction des 4 mutations engendrées artificiellement ?
Les 3 mutations engendrées artificiellement (par génie génétique) sur le virus concernent une région de la protéine de surface, l’hémagglutinine (HA) de type H5 qui interagit avec un récepteur cellulaire d’entrée dans les organismes des volailles par l’intermédiaire du récepteur α 2,3 — lié SA (acide sialique) présent de façon prédominante dans leurs poumons et leur intestin. Ce récepteur est très similaire au récepteur α 2,6 — lié SA, qui est plus spécifique aux poumons de l’être humain et du furet, même si le récepteur α 2,3 — lié SA est également présent chez l’homme et le furet, ce qui en fait un hôte intermédiaire.
De plus, ces mutations d’adaptation au récepteur humain α 2,6 — lié SA ne suffisent pas à rendre le virus transmissible par aérosol ou gouttelettes respiratoires. Il faut pour cela d’autres modifications des protéines de surface de virus que l’on peut obtenir par sélection successive. Il faut forcer l’infection nasale des furets, les laisser développer le virus, les euthanasier, récupérer les virus répliqués dans leurs poumons et leur gorge et réinfecter d’autres furets avec la solution super concentrée en virus (solution surnageant), et répéter l’opération (10 fois en l’occurrence) jusqu’à ce que le virus acquière de lui-même par mutation la capacité de devenir transmissible par voie aérienne.
Ron Fouchier n’a pas fait preuve d’un génie intellectuel hors norme pour concevoir les mutations spécifiques qu’il a introduit dans le génome de la souche sauvage du virus H5N1 aviaire pour le rendre transmissible entre mammifères. Ce n’est pas le fruit d’idées géniales ni le fruit du hasard. Non, il a tout simplement utilisé toutes les connaissances déjà solidement établies sur la façon dont les volailles se contaminent entre elles via l’interaction entre la protéine de surface H5 et les récepteurs SA qui se trouve dans leur intestin et leur poumon. La structure du site d’interaction entre H5 et les récepteurs α 2,3 — lié SA et potentiellement α 2,6 — lié SA est connue depuis 2006 par cristallographie par rayon X.
Dans son article, Ron Fouchier met un point d’honneur certain à décrire de façon exhaustive avec moult détails, comment ce genre de forçage d’adaptation peut être obtenu sélectionnant ainsi une 4e mutation spécifique de la protéine H5 qui permet au virus H5N1 muté d’être transmissible par voie aérienne entre furets. En tant que scientifique, en lisant l’article de Ron Fouchier ce qui frappe c’est le niveau et la rigueur des descriptions dans la précision des méthodes utilisées. Cela va bien au-delà du standard des articles habituels où souvent il faut être soi-même un expert pour comprendre les détails du mode opératoire. L’article de Ron Fouchier est une clé donnée au monde pour fabriquer facilement des virus de grippe A hautement pathogènes et transmissibles entre êtres humains.
Quelle est la probabilité que ces mutations arrivent naturellement ?
En effet, calculer la vitesse à laquelle un virus mute est une tâche importante pour l’établissement des vaccins, délicate, mais pas impossible, en particulier pour les virus de grippe A où il y a suffisamment de données accumulées. Pour la grippe A humaine le taux a été établi en 1991 à 6.7 x 10-3 mutations de nucléotide par site mutable et par par an pour le gène HA. En ce qui concerne le virus H5N1, le taux de mutation établi depuis 2011 se trouve entre 4 et 9 x 10-3 mutations de nucléotide par site et par an selon les sous-types de souches.
Il faut compter que seulement 50 % de ces mutations de nucléotides correspondent réellement à un changement au niveau de la protéine exprimée. Donc, a priori la probabilité d’obtenir une mutation naturelle simultanée des 4 sites transformés par Ron Fouchier est, en prenant le taux le plus élevé, de 0.5 x (9 x 10-3) à la puissance 4, c’est-à-dire environ 4 10-10 par an, donc moins d’une chance sur 1 milliard par an ! Et pour obtenir spécifiquement les 4 mutations sélectionnées il faudrait encore abaisser cette probabilité, car pour chaque site muté il peut correspondre plusieurs acides aminés possibles viables chez les volailles sans pour autant pouvoir affecter l’être humain.
Nous voyons donc qu’il y a au moins 3 fois plus de chance de gagner le jackpot à un tirage de l’EuroMillions que de chances de voir le virus H5N1 muter naturellement vers la forme pandémique pour l’homme en une année. Sur cent ans la probabilité reste encore de moins d’une chance sur 10 millions !
partie2 — Le virus synthétique hybride de Shi Zheng Li et Ralph Baric, une étape de plus franchie vers un monde nouveau d’insécurité biologique
Shi Zheng Li utilise un ressort de peur similaire, même si elle évoque un processus de recombinaison (mixing génétique) au lieu d’un simple processus de mutations en des points particuliers du génome.
Dans ce processus, des pans entiers de génomes sont échangés entre deux virus de façon naturelle créant un nouveau virus apparenté aux 2 précédents qui se sont recombinés en 2 autres virus différents en échangeant des parties de leurs génomes. Dans la nature ce procédé se limite à des organismes en principe très voisins. Mais avec le génie génétique en peut très bien introduire n’importe quel gène dans n’importe quel organisme (ex. gène de méduse fluorescente dans le mouton) et réfléchir ensuite à ce qu’on a fait.
Les coronavirus de chauves-souris pourraient très bien franchir la barrière des espèces en se recombinant entre eux pour ensuite affecter l’être humain. Et donc ils ont décidé d’en créer un en laboratoire pour prouver que c’est une réalité sur laquelle il faut compter et s’y préparer. En réalité, Shi Zheng Li a découvert et explique au monde entier quelque chose de bien plus inquiétant que ce qu’elle prétend en attirant l’attention sur les recombinaisons potentielles naturelles de coronavirus de chauves-souris. Ce sont les recombinaisons synthétiques à gain de fonction qui vont être le plus à craindre qui, d’ailleurs, potentiellement ne seront pas limitées à des virus de la même famille.
Description de la recherche de Shi Zheng Li à l’institut de virologie de Wuhan en collaboration avec Ralph Baric à l’Université de Chapel Hill en Caroline du Nord
France Soir a été plus que vivement critiqué par NewsGuard, un site anti-complotiste américain qui se targue de poursuivre et dénoncer les fake news et décerne des labels de qualité aux médias de la presse écrite. France Soir avait reçu un label rouge infamant au titre, en autre, d’un article sur un virus COVID synthétique, hautement pathogène pour l’être humain, qui avait été fabriqué dès 2015 par Shi Zheng Li en collaboration avec Ralph Baric (Cornell University, Caroline du Nord, USA). Selon News Guard, France Soir tentait de laisser penser au lecteur que le virus de la COVID-19, le SARS-Cov2, était une fabrication synthétique. Nous n’avons jamais écrit cela dans notre article, mais simplement informé le public qu’un autre virus de type COVID provoquant le SARS, artificiel et potentiellement hautement pathogénique pour l’être humain avait été créé en 2015 en laboratoire sans que jamais le grand public n’en ait été informé.
Chez France Soir, nous pouvons nous tromper, mais nous sommes sérieux. Pour le prouver et que le lecteur puisse juger par lui-même, nous retranscrivons sans modifications, ci-dessous, les éléments essentiels tirés de l’article de 2015 paru dans Nature Medicine. Les quelques commentaires que nous ajoutons n’ont pour but que de permettre au lecteur de comprendre le jargon technique (nous avions également traduit l’article dans son intégralité).
Il est très important de comprendre que le virus COVID synthétique a été réalisé en fait dans le laboratoire de Ralph Baric à l’Université de Chapell Hill dont le niveau de sécurité est seulement P3. L’équipe de Shi Zheng Li a contribué de façon décisive à cette recherche, car elle en a fourni l’élément essentiel, c’est-à-dire le gène de la protéine spicule SHC014, clé d’entrée dans les cellules pulmonaires humaines par l’intermédiaire du récepteur ACE2, qui a été intégré dans le génome du SARS du 2003 à Chapel Hill sous la responsabilité de Ralph Baric. Cette réalisation de ce premier virus COVID GOF hautement pathogène est incontestablement un fruit 100 % sino-américain. Nous aborderons dans le prochain chapitre les raisons qui font que cette réalisation n’a pas été faite intégralement à Wuhan alors qu’elle correspond à la mise en pratique d’une théorie développée par Shi Zheng Li depuis 2004.
Voici ce que Shi Zheng Li et Ralph Baric écrivent :
Écrit essentiel 1 : « Ici, nous examinons le potentiel pathogénique d’un virus de type SARS, SHC014-CoV, qui circule dans les populations de chauves-souris fer à cheval chinoises [1]. En utilisant le mécanisme de transcription inverse du SARS-CoV, nous avons créé et caractérisé un virus chimérique qui exprime la protéine de surface S SHC014 (“spicule”) d’un coronavirus des chauves-souris sur la structure de base d’un virus SARS-CoV adapté aux souris. »
Explication 1: Le SHC014-CoV est un coronavirus de chauves-souris dont la protéine de surface (dite spicule ou protéine pointe) spécifique porte le nom SHC014. Les coronavirus sont des virus à ARN et ne contiennent donc pas d’ADN. La transcription inverse est le mécanisme par lequel l’ARN contenu dans le virus va se transformer en ADN viral qui va être inséré dans l’ADN de la cellule infectée. Ici, en fait, il s’agit juste de recréer artificiellement l’ADN du virus SARS-CoV humain (celui de l’épidémie de 2003) afin d’y substituer par génie génétique sa protéine S avec celle du SHC014-CoV de chauves-souris. Elle indique une chose très importante, c’est que le virus humain SARS-CoV de 2003 a été adapté aux souris. Cela a une énorme importance, car cela veut dire que le virus a en principe perdu son caractère pathogénique pour l’homme. L’article ne dit pas comment, mais le virus a été adapté aux souris par une méthode qui s’apparente à celle décrite dans la partie 1 de l’article, concernant la recherche de Ron Fouchier.
Shi Zheng Li a donc créé un virus viable, hybride (chimérique) entre le SARS-Cov de 2003 adapté aux souris et portant la protéine S (SHC014) provenant d’un coronavirus de chauves-souris fer à cheval, lui-même identifié par elle-même et son équipe, en 2013 au laboratoire de virologie de Wuhan (référence [1]).
La grande particularité du virus SHC014-Cov, première découverte majeure de Shi Zheng Li en 2013, était sa capacité à infecter les humains par l’intermédiaire supposé du récepteur d’entrée cellulaire ACE2, mais également d’autres espèces mammifères porteuses de variants génétiques (appelés orthologues) de ce récepteur comme les chauves-souris fer à cheval et la civette palmiste, l’hôte intermédiaire de la pandémie de 2002-2003.
Le but de la recherche de Shi Zheng Li en 2015 était de confirmer l’hypothèse déjà hautement probable que l’infection du SHC014-Cov se faisait bien par l’intermédiaire du récepteur ACE2. Dans le reste de l’article, elle se réfère au SARS-CoV chimérique (virus hybride formé) comme le SHC014-MA15, c’est-à-dire le coronavirus de l’année 2015 adapté aux souris et portant la protéine spicule SHC014.
Écrit essentiel 2 : « Malgré les prédictions de modélisation de structure et les expériences de pseudotypage, le virus hybride SHC014-MA15 s’est révélé viable et se répliquait à de forts niveaux de concentration in vitro dans les cellules de type Vero. »
Explication 2 : cela veut dire que l’étude par modélisation structurale de la protéine SHC014 ne permettait pas de conclure qu’elle pouvait interagir avec le récepteur ACE2 humain. De même les expériences de pseudotypage, qui consistent à fabriquer un pseudo virus (avec une enveloppe protéinique n’étant pas celle du SARS-Cov) et portant la protéine SHC014, ne permettaient pas non plus la liaison au récepteur ACE2. Par contre, le virus hybride complet était viable et se répliquait donc dans les cellules du type Vero. Ces dernières sont des cellules hôtes pour la culture de virus qui permettent de mesurer, par exemple, la réplication de virus en présence ou en absence de médicaments expérimentaux.
Écrit essentiel 3 : « Ces virus se répliquent avec efficacité dans les cellules des voies respiratoires supérieures [sous-entendu humaines, NDLR] en atteignant des concentrations in vitro comparables aux souches épidémiques du SARS-CoV. »
Explication 3 : dans le résumé de l’article, quasiment le seul élément de texte lu par les scientifiques qui ne sont pas directement experts dans le domaine, Shi Zheng Li omet habilement de spécifier qu’il s’agit des voies respiratoires humaines !… En clair, le virus hybride créé se répliquait dans les cultures cellulaires (in vitro) des voies respiratoires humaines à un niveau de concentration mortel identique à celui mesuré chez les patients de 2003.
Dans le corps l’article cela est dit clairement :
Écrit essentiel 4 : « Pour tester la capacité de la protéine SHC014 de permettre l’infection des voies respiratoires humaines, nous avons examiné leur sensibilité a ce virus en utilisant la lignée cellulaire Calu-3 2B4 (réf. 9). Nous avons observé la capacité de ce virus à les infecter et à produire une réplication robuste du SHC014-MA15, comparable à celle du SARS-CoV Urbani (Fig. 1c). [il s’agit du virus de SARS de 2003, NDLR] »
Écrit essentiel 5 : « De plus, les expériences in vivo démontrent la réplication du virus chimérique dans les poumons des souris avec un effet pathogénique notable. L’évaluation des thérapies immunes contre le SARS [le syndrome respiratoire aigu sévère, NDLR] et des modalités prophylactiques montre peu d’efficacité ; que ce soit par l’intermédiaire d’anticorps monoclonaux ou de vaccins il n’a pas été possible de neutraliser l’infection des virus chimériques CoVs portant cette nouvelle protéine S. »
Explication 5 : cela se passe de commentaire !
Écrit essentiel 6 : « … nous avons vacciné les souris âgées avec le virus complet du SARS-CoV doublement inactivé (DIV) »
Explication 6 : une méthode puissante de vaccination est par virus doublement inactivé (DIV), c’est-à-dire tué en principe deux fois, pour garantir qu’aucune particule virale résiduelle ne reste et ne soit capable de se reproduire dans l’organisme. De tels vaccins sont proposés pour le COVID-19 (SARS-Cov2). Par contre l’aluminium sera systématiquement utilisé comme adjuvant (booster immunitaire), car développer un vaccin avec un autre type d’adjuvant sans aluminium nécessiterait probablement des recherches beaucoup plus longues.
Écrit essentiel 7 : « … cependant, le vaccin a échoué à protéger les animaux âgés qui présentaient également une pathologie immune, indiquant la possibilité que leur état ait été aggravé à cause de la vaccination. »
Explication 7 : en effet, tout vaccin peut engendrer à différents degrés des réactions auto-immunes. Ici, il semblerait que ces réactions soient assez fréquentes puisque seulement quelques douzaines de souris ont été testées !
Écrit essentiel 8 : « L’ensemble de ces observations confirme que le vaccin par virus atténué ne protégerait pas contre une infection par SHC014 et pourrait probablement augmenter la maladie dans un groupe vaccine plus âgé [ici les auteurs implicitement parlent des êtres humains ! NDLR]. »
Explication 8 : les vaccins atténués diffèrent des vaccins inactivés par le fait que le virus n’est pas complètement tué et garde une activité résiduelle. C’est cas de nombreux vaccins comme celui le BCG ou plus récemment celui du ROR. Les vaccins atténués ne peuvent pas absolument pas s’utiliser contre des personnes dont le système immunitaire est affaibli, car ils peuvent conduire, comme il est malheureusement souvent observé, au développement de la maladie au lieu de l’empêcher.
Écrit essentiel 9 : « Ces résultats démontrent que des anticorps largement neutralisants contre le SARS-CoV n’ont qu’une efficacité marginale contre des souches émergentes de virus du type SARS-CoV comme le SHC014-CoV. »
Explication 9 : il n’y a pas d’immunité croisée avec le SARS-Cov de 2003 !
Écrit essentiel 10 : « Nous considérons les virus portant la protéine SHC014 comme une menace potentielle à cause de leur capacité de se répliquer dans les cultures de cellules des voies respiratoires supérieures humaines, le meilleur modèle à disposition pour l’étude des maladies respiratoires. De plus, la pathogénie observée chez les souris indique une capacité pour les virus contenant la protéine SHC014 de causer une maladie dans les modèles mammifères sans adaptation du RBD [RBD : domaine de liaison de la protéine spicule SHC014 au récepteur d’entrée cellulaire ACE2, NDLR]. »
Explication 10 : il faut normalement une adaptation par mutation ou par recombinaison naturelle, dans un ou plusieurs hôtes intermédiaires, pour qu’un virus franchisse la barrière des espèces vers l’homme. Ici, ce n’est pas le cas, le seul ajout de la protéine SHC014 suffit ! C’est une découverte qui laisse planer un danger sur l’humanité.
Les peurs du Haut Conseil pour la Santé Publique découlent de cette publication sur laquelle l’omerta règne
À la lecture détaillée de la recherche GOF de Shi Zheng Li, on se rend compte pourquoi le gouvernement a si peur de l’épidémie de COVID-19 et pourquoi les bruits les plus anxiogènes se sont répandus dans la presse dès le début de la pandémie, comme sur l’impossibilité supposée de la vaccination et plus tard sur l’absence d’immunité croisée. Le haut conseil pour la santé publique se référait de toute évidence à la publication de Shi Zheng Li sans en informer la presse et donc le public. Celui-ci n’avait visiblement pas le droit de savoir qu’un coronavirus pouvant causer le syndrome respiratoire SARS (donc un virus COVID) avait été synthétisé en laboratoire déjà depuis 2015, mettant en avant des difficultés potentielles à le combattre.
Il faut noter également que les contradictions apparentes, dans les médias, sur les délais de mise au point d’un vaccin proviennent également de ces observations initiales faites par Shi Zheng Li et son équipe. Établir un vaccin fiable destiné à un usage très large est toujours une tâche difficile qui peut prendre plusieurs années parfois. Dans des cas extrêmes, il peut s’avérer impossible pendant des décennies de mettre au point un vaccin, comme cela est le cas pour le virus du SIDA. Un des problèmes possibles dans la mise au point d’un vaccin est la recherche d’un booster immunologique (adjuvant) efficace. Parfois, cela prend énormément de temps de le mettre au point. Cependant, utilisation de sels d’aluminium comme adjuvant permet préparer presque à coup sûr un vaccin qui serait autrement bien plus difficile à réaliser.
Il faut noter, en a parte, qu’à propos du débat sur la sécurité des vaccins, l’aluminium est un poison absolu pour les êtres vivants et que le système immunitaire met en œuvre une réaction de rejet contre lui telle que les chances d’obtenir en même temps des anticorps contre les fragments du virus présents dans le vaccin sont plus que décuplées. Injectée dans le muscle une toute petite fraction de l’aluminium finit par atteindre le système nerveux central et s’y accumuler dans les neurones, vaccination après vaccination. La limite de tolérance du nombre de vaccinations dépend de la variabilité physiologique entre individus. Le syndrome dû aux sels d’aluminium (myofasciite a macrophages) présent dans les vaccins a été démontré.
Ce dont les autorités françaises devraient avoir vraiment peur c’est que la découverte faite par Shi Zheng Li permettrait de rendre n’importe quel coronavirus hautement pathogénique pour l’homme, sans nécessité d’adaptation par recombinaison ou mutation dans un hôte intermédiaire, en y substituant sa protéine spicule avec celle du virus SHC014-Cov de chauves-souris.
Quelles conclusions en tirer ?
Les apprentis sorciers ne manquent pas d’hypocrisie sur l’utilisation duale de leur recherche
Ce qui peut apparaître finalement comme le nec plus ultra de l’inconscience, c’est que ces apprentis sorciers exposent au monde entier leurs expériences tellement ils en sont fiers. En les rendant ainsi publiques, ils concourent doublement au franchissement de la barrière des espèces : d’une part par le risque de fuite à l’extérieur des laboratoires des virus qu’ils créent et, d’autre part, en livrant des recettes opérationnelles aux autres chercheurs au service d’états peu scrupuleux ou d’organisations terroristes non étatiques. Il est en effet beaucoup plus facile de reproduire une recette que de la mettre au point et les moyens à mettre en œuvre peuvent être bien plus réduits, surtout en termes de précautions. Les laboratoires P4 ne sont pas une obligation et les kits de mutation ciblée (mutagenèse dirigée) peuvent s’acheter couramment.
En réalité, les chercheurs impliqués dans les recherches GOF sur les virus ne manquent pas de lucidité. Ils sont parfaitement au courant de l’usage dual potentiel de leurs découvertes. En particulier, Shi Zheng Li ne pouvait pas ne pas être au courant du programme militaire chinois d’identification et d’étude des coronavirus de chauves-souris, programme initié et conduit en parallèle à celui des universitaires depuis le début des années 2000. Nous consacrerons le prochain chapitre de l’histoire du Covid-19 au programme militaire chinois.
Les scientifiques dont nous avons décrit les recherches GOF sont donc responsables de leurs actes. Ils sont très mal à l’aise avec cela, car ils n’évoquent tout simplement jamais l’usage dual qui peut être fait de ce qu’il considère être des avancées dans la lutte contre les pandémies. Des pandémies qui, précisons-le, n’existent pas et dont les chances d’émergence naturelles sont infiniment faibles comme nous l’avons vu pour le H5N1 muté de Ron Fouchier, mais également pour l’hybride de SARS-Cov que Shi Zheng Li a fabriqué. À force de vouloir éviter au monde des fléaux, il semblerait qu’ils contribuent à accélérer le mouvement vers leur réalisation.
Ainsi les scientifiques ont dorénavant le champ libre, car interdire les recherches GOF est à présent tout aussi illusoire que d’interdire leurs publications comme le suggérait Lynn C. Klotz, l’experte américaine de l’institut Center for Arms Control and Non-prolifération, dans son rapport tout en admettant d’ailleurs que c’est un vœu pieu.
Dans sa métaphore du chat qui s’est échappé du sac pour le contenir, elle nous indique qu’il n’est déjà plus possible d’empêcher la prolifération de création de PPP (virus pathogènes à potentiel pandémique). Dans son analyse au sujet de la recherche de Ron Fouchier, elle écrit : « les méthodes pour créer des virus transmissibles de façon aérienne sont bien établies et peuvent être reproduites par des chercheurs peu doués dans le domaine de la virologie moléculaire. » Son rapport sur les activités de Shi Zheng Li n’est pas encore rendu public.
En tout cas, depuis l’émergence du SARS en 2003, il y a eu six départs d’épidémie de SARS provenant de laboratoires de recherche, dont 4 en Chine. Ces fuites de laboratoires ont causé l’infection de 13 personnes dont une est morte (Furmanski, 2014). En réponse, les États-Unis ont banni certains types d’expérience GOF avec des PPP en 2014. Mais l’interdiction (un moratoire sur le financement) qui n’a jamais été appliquée (voir chapitre 2) a été levée en 2017.
En février 2019, dans le Bulletin of The Atomic Scientist, Lynn C. Klotz conclut que les erreurs humaines sont à l’origine aux USA de la plupart des accidents de laboratoires de haute sécurité causant l’exposition aux pathogènes. Alors que des problèmes d’ordre matériel étaient un facteur dans les 749 incidents rapportés aux autorités, entre 2009 et 2015, 79 % résultaient d’erreurs humaines.
Nous ne pouvons cesser de penser à cette virologue française, interviewée au mois d’avril 2020 par un grand quotidien, qui assurait la main sur le cœur qu’aucun chercheur chinois n’aurait osé faire des manipulations génétiques sur les virus du SARS !
Alors, s’il vous plaît les apprentis sorciers, arrêtez votre déni et assumer vos responsabilités, car vous nous faites penser au Tartufe de Molière et sa fameuse tirade : « Cachez, Madame, ce sein que je ne saurais voir ! ».
chapitre 4
partie 1 — Les virus de Zhoushan

Chauve-souris (Chine, musée du Quai Branly) Crédit : Jean-Pierre Dalbéra
Nous tentons dans ce chapitre de donner un éclairage sur la réalité de l’activité militaire chinoise en matière de recherche sur les coronavirus de chauves-souris. Nous nous concentrons ici sur la Chine puisqu’il s’agit de l’histoire d’un virus pandémique qui a émergé du sol de cette grande puissance.
Partie 1. Position de la Chine sur l’échiquier géopolitique des grandes puissances
La Chine vise la dominance de façon peu discrète
Par sa superficie et sa population, mais aussi par son développement économique hégémonique et technologique récent, tout à fait hors norme, la Chine semble avoir retrouvé son statut d’Empire du Milieu. Pour certains, la signification taoïste hautement symbolique d’un centre du monde, situé à la conjonction immatérielle du Ciel et de la Terre, a fait place au concept pragmatique d’axe de révolution économique du monde post-moderne. Ces caractéristiques la font apparaître aux yeux de nombreux experts en géopolitique comme le seul challenger possible des USA pour la position de première puissance mondiale. Ce statut géopolitique construit en quelques décennies seulement a des répercussions considérables sur la vie économique et sociale de l’ensemble de la planète. Cette domination économique s’accompagne d’une affirmation géopolitique de grande puissance qui, pour ne pas être contestée, doit s’appuyer parallèlement sur une souveraineté militaire permettant de faire face aux USA. La Chine revendique d’ailleurs sa souveraineté sur Taïwan et souhaiterait expulser les Américains de l’île de Guam où ils détiennent une base militaire nucléaire stratégique pointée vers la Chine.
En réalité, la Chine ne détient pas encore complètement la domination géopolitique face aux USA, la seule puissance qui peut sans être contestée, politiquement et militairement, porter la guerre quasiment n’importe où dans le monde, en dehors de la Chine, la Corée du Nord (protégée par la Chine) et la Russie.
Dans les années 1980, le leader chinois Deng Xiaoping avait dit : « Observer calmement ; garantir nos positions ; traiter nos affaires calmement ; dissimuler nos capacités et attendre notre heure ; maintenir un profil discret et ne jamais revendiquer le leadership. » (source : Le Figaro).
Nous pouvons à présent mesurer le chemin parcouru par la Chine sur le plan géopolitique par rapport à cette citation historique où elle endosse officiellement, face aux USA, le rôle de l’underdog : le chien qui ne mène pas la meute et doit se soumettre en attendant de pouvoir un jour prendre un rôle de leader grâce à ses qualités propres. En ce qui concerne leur capacité dans le domaine des virus GOF, il semblerait qu’ils ne peuvent plus faire profil bas si l’on en juge par les recherches publiées de Shi Zheng Li à l’institut de virologie de Wuhan.
La Chine a-t-elle manipulé les USA ?
La Chine est-elle devenue transparente ? Ou bien faut-il comprendre que les recherches publiées par Shi Zheng Li ne sont que la partie émergée d’un iceberg de recherches GOF en Chine. Elles pourraient être conduites dans d’autres laboratoires plus confidentiels comme le P4 de l’institut vétérinaire de Harbin ou par l’université militaire de recherche de Zhoushan (voir partie 3 de ce chapitre).
La recherche GOF que nous avons décrite dans le chapitre précédent impliquait une collaboration étroite avec les USA, ce qui présentait de multiples avantages. Elle permettait l’acquisition de technologie en même temps qu’elle garantissait une couverture morale basée sur l’affirmation que la Chine ne faisait rien en secret dans de domaine de recherche très sensible. Dans tous les domaines, la Chine est experte dans les stratégies d’acquisition de technologies. Elle y arrive parfaitement en tenant une place en apparence effacée.
Dans sa collaboration avec le laboratoire de Ralph Baric à Chapel Hill, Shi Zheng Li a fourni le gène de la protéine SHC014 certainement contre l’acquisition d’une technique de pointe en manipulation génétique, probablement par l’intermédiaire d’un chercheur postdoctoral envoyé se former dans le laboratoire de Ralph Baric. Lorsque l’on étudie la publication de 2015 relatant la fabrication d’un virus COVID synthétique, seuls les noms de Shi Zheng Li et Xing-Yi Ge, l’étudiante ou collaboratrice postdoctorale de Shi Zheng Li, qui avait signé en premier auteur (dans Nature en 2013) la découverte du virus SHC014-Cov, sont chinois. Parmi les 13 autres auteurs, 9 sont affiliés à l’Université de Chapel Hill, dont le premier et le dernier auteur qui sont les plus importants dans une publication scientifique. On trouve également 2 auteurs affiliés au Department of Cancer Immunology and AIDS du Dana-Farber Cancer Institute (Harvard Medical School, Boston, Massachusetts), 1 auteur affilié au National Center for Toxicological Research (Food and Drug Administration, Jefferson, Arkansas) et 1 auteur de l’Institute for Research in Biomedicine de Zurich (Bellinzona Institute of Microbiology). Tous ces instituts sont des centres de recherche nord-américains et européens de première classe.
Cet étonnant attelage contre nature permettait à chacun de tirer parti de l’autre tout en pensant jouer au plus fin et en se réservant la possibilité d’accuser l’autre en cas de faute. En effet, nous avons vu au début de la pandémie qu’Américains et Chinois, en bons frères ennemis, se sont accusés mutuellement d’en être à l’origine.
Stratégies des autres grandes puissances face aux recherches GOF sur les virus
Comme nous l’avons écrit au chapitre 2, les USA, la Russie et la Chine ont de toute évidence eu dans le passé des programmes militaires secrets dans le domaine des virus pathogènes. Mais aucune information ne transparaît sur les recherches possibles que ces puissances pourraient développer à l’heure actuelle. En tout cas, les USA ont développé une veille militaire semi-officielle sur les recherches GOF conduites sur les virus dans les laboratoires universitaires civils aux USA et en Asie tout en leur laissant la liberté d’y procéder.
D’autre part, le soin particulier et la rapidité avec lequel la Russie s’applique à développer depuis quelques années des vaccins contre les virus les plus pathogènes, comme ceux de l’Ebola, du Zika (chikungunya) et à présent du Covid-19, indiquent le souci réel que représentent les virus pandémiques aux yeux des stratèges politiques et militaires russes qui veulent montrer au monde qu’ils s’y préparent activement. L’ancien centre secret soviétique d’élaboration d’armes bactériologiques, reconverti en centre de recherche en biotechnologie et virologie, en témoigne. Baptisé du nom évocateur Vektor il est situé près de Novossibirsk en Sibérie occidentale. C’est un lieu névralgique de la recherche vaccinale russe. « Ce laboratoire a été agréé pour être l’un des deux seuls laboratoires au monde, avec le Centers for Disease Control and Prevention (CDC) à Atlanta (États-Unis), à détenir le virus de la variole » (source 20 Minutes).
La presse française, contrôlée par l’oligarchie financière internationale qui relaie les intérêts euro-atlantistes anti-Poutine, met un point d’honneur à systématiquement dénigrer, sans avancer le moindre élément de preuve, l’efficacité des vaccins russes. Plus récemment cependant, un article de La Tribune met en avant l’efficacité du vaccin russe Spoutnik V démontrée dans un article publié par le Lancet ! La presse devrait plutôt analyser les raisons qui poussent les Russes à développer des vaccins contre des infections, comme les virus Ebola et Zika, qui a priori ne les concernent pas. Par exemple, un article de SlateFr met en avant que malgré le fait que le vaccin Spoutnik V contre le SARS-Cov2 n’ait été testé que sur une quarantaine de personnes il pourrait être administré à l’intégralité de la population russe. Il y a de quoi sourire ! SlateFr est dans la parabole de la paille et la poutre. D’une part il s’agit là d’une affirmation gratuite et, d’autre part, c’est exactement ce que prévoient de faire certains décideurs au sein des gouvernements occidentaux (voir chapitre 3 partie 1).
Rappelons, en a parte, que tous les vaccins présentent des effets délétères de différentes natures et à des degrés variables (chapitre 2 et chapitre 3 partie 2). Les réactions immunitaires qui dégénèrent en syndrome auto-immun (ex. syndrome de Guillain Baré…) sont en principe rares avec quelques cas observés sur des centaines de milliers, de vaccinations. Cependant certains vaccins se révèlent plus nocifs que d’autres et lorsque l’on envisage une vaccination de masse il faut être sûr du niveau de sécurité du vaccin. Un exemple de ratage peu mis en avant par la presse est l’effet débilitant observé du vaccin Gardasil contre le virus du papillome humain. Au 9 septembre 2018, au total 59.634 cas de réactions adverses au Gardasil avaient été enregistrés depuis l’ouverture du site internet de l’association Sane Vax aux USA. Dans 15.390 cas, les jeunes femmes se sont retrouvées aux urgences ; 5.952 ont dû être hospitalisées dont 994 dans un état engageant le processus vital ; 438 sont décédées et 15.000 ont gardé une invalidité permanente ou ne se sont pas véritablement remises.
La doctrine russe durant la guerre froide était basée sur la « réponse du mort » illustrée par le célèbre film de Stanley Kubrick « Docteur Folamour » qui démontrait comment la folie humaine pouvait conduire à la destruction de la planète. La version OTAN du concept de la réponse du mort s’appelle MAD (Mutually Assured Destruction ou destruction mutuelle assurée en français) un acronyme d’un cynisme particulièrement savoureux.
Poutine a récemment réaffirmé qu’en cas de conflit nucléaire, avec menace de lancers de missiles nucléaires balistiques elle ne frapperait pas en premier, mais que de toute façon il n’y aurait pas de gagnant ni de survivant d’ailleurs! À présent, pour sanctuariser son sol et sa population, elle prévoit également la réponse à des conflits de conception plus moderne avec l’usage d’armes nucléaires tactiques (c’est-à-dire à portée limitée sur des théâtres d’intervention réduits), mais aussi la possibilité qu’elle soit attaquée par d’autres types d’armes de destruction massive qui engageraient ses intérêts vitaux ! Évidemment, il s’agit là d’une référence implicite à des armes bactériologiques dont les virus exterminateurs de type Ebola ou de l’épidémie de SARS de 2003 font partie.
Il n’est pas impossible que la Russie prévoie avec sagesse qu’une attaque par un virus pandémique militaire pourrait être parfaitement déguisée en virus naturel (comme nous le verrons dans un prochain chapitre) et que dans ce cas la seule défense acceptable serait de pouvoir établir un vaccin de toute urgence pour éviter au monde la réponse du mort ! Avec le Covid-19, nous sommes entrés dans une ère géopolitique tout à fait nouvelle, d’insécurité mondiale, et nous ne connaissons pas encore qu’elle sera la réponse diplomatique et politique du reste du monde face à la Chine ? Cependant, il n’est pas certain qu’il y en ait une, car la marge de manœuvre des USA et des pays de l’UE, de toute façon alignés sur les USA, semble très étroite. En tout état de cause, que la pandémie soit d’origine naturelle ou accidentelle, la Chine devrait rendre des comptes devant les instances de l’ONU sur le fait qu’après l’épidémie de SARS de 2003 ses marchés aux animaux sauvages ont été maintenus, sur la défaillance du système d’alerte prétendument mis en place, sur sa main mise sur l’OMS et sur ses activités de recherches GOF sur les coronavirus causant le SARS. L’absence de réponse officielle de sa part, ce qui est fort probable, validera sa domination géopolitique sur le reste monde.
En ce qui concerne la France, grande puissance nucléaire siégeant au Conseil de Sécurité de l’Organisation des Nations Unies, sa doctrine se limite à ce que nous avons expliqué au chapitre 2 (partie 2), c’est-à-dire un partenariat civil altruiste de dupe avec la Chine.
Partie 2 — Les chauves-souris — réservoir naturel ancestral des coronavirus infectant l’Homme
Les coronavirus comme tous les virus ont une origine phylogénique inconnue. C’est-à-dire que l’identification de leur ancêtre primordial dans l’évolution de la vie sur terre ne peut pas se faire, même approximativement, comme on a pu le faire pour nombre d’espèces animales grâce aux fossiles découverts dans les couches sédimentaires pétrifiées. Les virus microscopiques et sans squelette ne laissent pas de trace fossilisée dans les roches. Les quelques chances de trouver des traces de virus remontant à l’ère des dinosaures se trouvent dans des parasites conservés par miracle dans de l’ambre datant de plus de cent millions d’années. Par contre les éléments de machinerie cellulaire que les virus utilisent prouvent qu’ils ont existé de tout temps, la question étant de savoir s’ils sont apparus avant les cellules primordiales ou après. On peut retracer la présence ancestrale des rétrovirus grâce aux vestiges de leur ADN insérés dans les génomes d’animaux hôtes dont l’être humain, ce qui permet d’affirmer qu’ils existent et ont co-évolué depuis des dizaines de millions d’années avec leurs hôtes. Des recherches sur les génomes viraux indiquent la possibilité qu’ils puissent provenir d’une autre branche du vivant remontant aux cellules primordiales il y a plus de 3 milliards d’années.
Avec plus de 1100 espèces colonisant tous les continents hormis les régions circumpolaires, les chauves-souris, appelées aussi chiroptères, représentent un quart de toutes les espèces mammifères derrière les rongeurs dont elles descendent. Comme les rongeurs, elles sont apparues avant la plupart des autres mammifères dont l’ère a débuté il a environ 60 millions d’années. Elles ont connu une diversification évolutive rapide sans grands changements morphologiques (raccourcissement ou perte de l’appendice caudal). Les fossiles de chauves-souris sont datés entre 20 et 30 millions d’années, le plus ancien découvert récemment au Wyoming est daté à 50 millions d’années (Éocène, début de l’ère tertiaire). La domination des airs par leur type d’ailes est une réussite très précoce de l’évolution transmise aux mammifères comme en témoigne le fossile de dinosaure chauve-souris, datant de 165 millions d’années, découvert en Chine récemment.

À ce titre, et comme le montrent de nombreuses publications scientifiques récentes, il est probable que le réservoir ancestral des coronavirus chez les mammifères soit les chauves-souris. En raison de leur habitat hivernal dans des cavernes montagneuses reculées ou des mines désaffectées, les chauves-souris sont par ailleurs en contact étroit avec le monde des insectes qui cohabitent par centaines de millions avec elles, se nourrissant de leurs déjections accumulées au fond des grottes. À cette occasion, la barrière des espèces entre insectes et mammifères peut être franchie par recombinaison virale. L’équipe du Pr Raoult de l’IHU de Marseille affirme avoir repéré une petite séquence de génome (transposon) de virus d’insecte intégré dans le SARS-Cov2.
Ces animaux au cours de leur évolution ont acquis une résistance particulière aux virus qui les infectent et elles en véhiculent autant que les rongeurs qui furent pourtant les premiers mammifères à apparaître sur terre et comptent 2 fois plus d’espèces. Plus de 130 espèces de virus ont été détectées chez les chauves-souris, dont 61 sont des virus de zoonoses hautement pathogéniques pour l’être humain. Cette spécificité contribue à prouver qu’elles sont le réservoir de coronavirus chez les mammifères depuis des temps immémoriaux. Les rongeurs eux ne véhiculent pas de coronavirus, mais des hantavirus qui peuvent également infecter l’être humain, mais de façon assez rare et peu contagieuse, créant des fièvres hémorragiques et des symptômes grippaux et respiratoires.
Une étude faite sur des rats en Chine a suggéré que certains beta-coronavirus (de lignée A) pourraient avoir émergé à partir de rongeurs. Le coronavirus (ChRCoV) HKU24, identifié chez le rat norvégien en Chine aurait divergé d’un ancêtre commun vers l’an 1400. Cette date très récente montre bien que les rongeurs ne sont certainement pas le réservoir des coronavirus affectant les mammifères, mais juste des hôtes intermédiaires potentiels dans le franchissement de la barrière des espèces.
Au contraire des rongeurs, les chauves-souris ont vécu pendant des millénaires dans un habitat séparé de celui des hommes qui n’ont jamais été confrontés à une large échelle au virus qu’elles véhiculent. Des virus remontant à la nuit des temps sont le fruit d’une très longue évolution dans ce réservoir. Ils sont parmi les plus terribles comme celui d’Ebola, le virus de Marburg ou les lyssavirus. Ces animaux nocturnes sont si discrets qu’on peut encore en redécouvrir de nouvelles espèces avec cet extraordinaire animal qui butine et féconde une fleur particulière (Anoura geoffroyi).
Premières zoonoses modernes transmises par les chauves-souris
Deux incidents de zoonoses transmises à l’homme avant l’an 2000 ont poussé les virologistes à se pencher sur l’étude approfondie des chauves-souris : l’émergence du virus Hendra, en Australie en 1994, passé des chevaux aux humains, et l’épidémie du virus Nipah, en Malaisie en 1998, issue des porcs. Dans le cas du virus Hendra douze chevaux d’un même élevage étaient morts. Le garçon d’écurie ainsi que l’entraîneur avaient été contaminés présentant des symptômes grippaux sévères. Le garçon d’écurie avait survécu tandis que l’entraîneur était décédé suite à des complications respiratoire et rénale. D’autres cas mortels de transmission à l’homme ont été rapportés depuis. Le virus Nipah est la cause d’une maladie neurologique et respiratoire mortelle touchant les élevages de porcs en Malaisie péninsulaire. Lors de son apparition en 1999, il a entraîné le décès de 115 personnes sur 265 affectées et l’euthanasie d’un million de porcs. L’enquête des infectiologues a démontré que les chauves-souris frugivores de la famille des ptéropidés (roussettes) étaient le réservoir de ces deux nouveaux Henipavirus mortels transmis à l’homme par l’intermédiaire d’animaux domestiques contaminés (Halpin et al. « Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus », Journal of General Virology, vol. 81, n° 8, 2000, p. 1927-32.) La destruction de leur habitat (déforestation en Asie du Sud-Est) ou l’empiétement de l’homme sur celui-ci est à l’origine de l’émergence de ces zoonoses dans l’environnement humain et permettant le franchissement des espèces.
Tout cela contribue au fait que ces animaux ont été, au moins à partir de la pandémie de SARS de 2002-2003, une cible de recherche privilégiée des universitaires chinois. Y avait-il un programme militaire conduit en parallèle dès 2003 ? Personne ne peut l’affirmer, mais il s’agissait bel et bien de débusquer des virus dangereux pour les chercheurs universitaires impliqués. « Les coronavirus transmis par les chauves-souris provoqueront d’autres épidémies », déclarait Shi Zheng Li en 2004 avec une certitude inquiétante, lors d’une campagne de prélèvement d’échantillons de sang de chauve-souris dans des zones reculées en Chine. « Nous devons les trouver avant qu’ils ne nous trouvent. » Elle contribuait dès 2005 à un article dans Science, intitulé : « Les chauves-souris sont le réservoir naturel des coronavirus de type SARS », qui laisse planer peu de doutes sur la réalité que nous décrivons dans le reste de ce chapitre.
Plusieurs années plus tard, au cours d’une surveillance longitudinale de l’espèce Rhinolophus sinicus (chauves-souris rousse chinoise dite « en fer à cheval ») dans la province du Yunnan, son équipe a réussi à isoler un coronavirus de type SARS (SL-CoV) extrait de cellules Vero E6 qui avaient été incubées dans leurs fèces, donnant lieu à la publication de 2013 qui a conduit par la suite à la réalisation du premier virus COVID synthétique en 2015 (chapitre 3). Le SL-CoV virus isolé en 2013 présentait plus de 95 % d’identité de séquence avec les virus SARS-CoVs humains et de civettes de l’épidémie de 2003. Il était capable d’infecter des animaux possédant des récepteurs orthologues (c.-à-d., des variants génétiques interespèces) du récepteur ACE2 humain (récepteur de pénétration cellulaire présent principalement dans les poumons), indiquant la possibilité d’infection directe des êtres humains sans recombinaison intermédiaire.
Dan Hu, un chercheur universitaire militaire chinois, dont les recherches sur les coronavirus de chauves-souris résument la situation mondiale ainsi : « Ces dernières années, de nombreux coronavirus ont été identifiés chez une large variété d’espèces de chauves-souris à travers le monde : en Asie, Europe, Afrique et Amérique. La plupart des coronavirus de type SARS (appelés SL-CoV) ont été identifiés à partir de chauves-souris en fer à cheval (rhinolophes) en Chine, Slovénie, Bulgarie et Italie. D’autres coronavirus proches du SARS-Cov de 2002-2003 ont été détectés dans les espèces Hipposideros et Chaerophon au Kenya et au Nigeria. »
Tous ces coronavirus de chauves-souris partagent entre 87 et 92 % d’identité de séquence avec le virus de SARS-Cov de 2002-2003. Les virus Rs3367 et RsSHC014 identifiés en 2015 par Shi Zheng Li, à la suite de sa campagne de surveillance dans le Yunnan, partagent plus de 95 % d’identité de séquence de génome avec le SARS-CoV isolé à partir d’êtres humains et de civettes. Cela démontre sans ambiguïté possible que les chauves-souris sont bien le réservoir naturel à partir duquel l’épidémie de 2003 a émergé.
Pour finir cette section sur une note de réflexion vaccinale ancienne. Comme nous l’avons écrit précédemment il est connu depuis longtemps que les chauves-souris peuvent engendrer des affections respiratoires. Dans la pharmacopée traditionnelle chinoise, la viande de chauves-souris permettrait de soigner l’asthme, ainsi que des affections rénales et systémiques. Il s’agit probablement de la mise en pratique du principe éternel de soigner le mal par le mal comme dans le concept de phàrmakon (substance qui contient à la fois le poison et l’antidote) des Grecs anciens.
Partie 3 — Le virus à l’origine de l’épidémie de 2002-2003 identifié dans une colonie de chauves-souris du Yunnan
En 2015, Shi Zheng Li écrivit, concernant les deux coronavirus particuliers qu’elle avait identifiés à la suite de sa campagne de surveillance effectuée en 2013 dans le Yunnan (dans une grotte située à 1 km d’un village à 60 kilomètres de Kunming) : « Le séquençage complet des génomes a révélé que les virus Rs3367 et RsSHC014 partageaient plus de 95 % d’identité avec les virus SARS-Cov humains et de civettes, ce qui était remarquablement plus élevé que l’identité de séquence de n’importe quel autre virus SL-Cov identifié (76 à 92 %). » Quel miracle ! Était-ce vraiment contre toute attente comme elle semble le suggérer dans sa publication ?
Pour les journalistes la question se pose encore de savoir comment le virus est passé de la grotte de Kunming aux marchés aux fruits de mer de Canton à quelque 1500 km de distance. Pour un esprit rationnel, cela signe indubitablement qu’un trafic de chauves-souris capturées dans le Yunnan a certainement alimenté le marché aux animaux sauvages de la ville de Canton située dans la province du Guangdong. Dans son article, Shi Zheng Li n’explique pas pourquoi elle a fait une campagne dans le Yunnan en 2013, mais il n’est pas interdit de penser qu’elle s’était renseignée sur les trafics de chauves-souris pour en remonter la piste. C’est en tout cas ce que tout chercheur scientifique aurait tenté de faire en suivant la logique d’un enquêteur. Il faut noter que ces marchés aux animaux sauvages dont les Chinois sont si friands se sont développés dans les années 1990, après la crise de la place Tiannamen en 1989, et la création d’un libéralisme radical par les autorités communistes. Ce fut l’avènement de la libre entreprise en Chine dans un esprit de business sans contrôle et de trafic en tout genre.
Il y a à présent en Chine des dizaines de milliers de marchés où s’entassent pêle-mêle des dizaines d’espèces d’animaux parfois réellement sauvages, comme des chauves-souris, ou très souvent du gibier d’élevage. Ces marchés peuvent s’étendre sur des surfaces géantes de plus de 100 hectares au sein de mégalopoles de plus de 10 millions d’habitants chacune. Ils constituent des bombes biologiques à retardement. Il n’a pas fallu 13 ans pour qu’une première zoonose de coronavirus émerge à Canton en octobre 2002. 17 ans après nous assistons impuissants à celle du SARS-Cov2, officiellement accrédité au marché aux animaux sauvages de Huanan à Wuhan. Ces marchés génèrent 100 milliards d’euros de chiffre d’affaires annuel et font vivre des millions de Chinois…
C’est un tel problème pour la Chine qu’elle n’a pas hésité à faire marche arrière en ce qui concerne le marché de Huanan avec l’annonce de Gao Fu, directeur du Centre chinois de contrôle et de prévention des maladies qui déclarait fin mai : « les prélèvements effectués là-bas (le marché de Huanan, NDA) début janvier n’avaient relevé aucune présence de virus sur les animaux. »(source CNEWS). D’ailleurs, CNEWS avec l’à-peu-près journalistique qui consiste à affirmer sans vérifier les informations se trompe dans son titre en affirmant que les premiers cas ont été détectés sur le marché aux fruits de mer. Au chapitre 1 nous avons montré qu’en réalité les premiers cas officiellement détectés en décembre 2019 n’avaient pas été en relation avec le marché de Huanan ou un des autres marchés annexes de ce type à Wuhan. D’autre part, des revues radiologiques rétrospectives ont montré que les tout premiers cas non détectés avaient eu lieu à la mi-novembre 2019 sans qu’aucune enquête n’ait eu lieu sur les lieux fréquentés par les patients ou les personnes avec lesquelles ils avaient été en contact.
En tout cas, cette déclaration de Gao Fu est l’inverse de celle qu’il avait faite 4 mois plus tôt, le 22 janvier 2020, quand il affirmait au reste du monde que le foyer de l’épidémie était le marché aux fruits de mer de Huanan. Devant un tel revirement, les gouvernements occidentaux restent paralysés et ne demandent toujours pas de compte à la Chine qui continue de se vautrer sans complexe dans la désinformation à répétition. On peut dire qu’elle aurait tort de se gêner. Cette dernière annonce n’a pour but ni la transparence ni la recherche de la vérité. Elle est venue à un moment où la thèse du pangolin, hôte intermédiaire, commençait à être sérieusement mise en doute sur le plan scientifique, par une publication parue le 14 mai dans PLOS Pathogène, une revue de prestige. Cet article semble avoir d’autant plus de poids qu’il est fait par des chercheurs chinois affiliés au Laboratoire Central de Préservation et d’Utilisation des Ressources Animales de Guangdong !
Nous verrons minutieusement au cours de chapitres suivants quel crédit on peut apporter à cette affirmation. Le mode de communication de la Chine sur les faits essentiels entourant le départ de l’épidémie à Wuhan étant, depuis le début, le fruit de manœuvres de désinformation telles que le fait que ce soit une équipe de chercheurs chinois qui démontre scientifiquement que ce n’est pas le pangolin le responsable, peut paraître suspect. Les autorités chinoises se sont-elles trouvées dans l’obligation d’adapter leur discours officiel ou bien ont-elles planifié une annonce qui leur permettait de noyer habilement le poisson en ce qui concerne le marché aux fruits de mer de Wuhan, et par conséquent ceux du reste de la Chine ? D’une part, elle laissait entendre que ce n’était pas la peine de fermer les marchés à risque puisque l’épidémie de Wuhan ne viendrait pas de là. D’autre part, elle laissait quand même planer le doute puisqu’elle avait fait procéder à la destruction des échantillons prélevés dont on ne sait pas si la liste incluait des chauves-souris ou pas.
De son côté, l’OMS reprend sans surprise la même explication comme le rapporte l’article de CNEWS : « il est même certain qu’il [NDA, le marché de Huanan] a joué un rôle. Mais quel rôle nous ne savons pas, que ce soit la source ou le cadre amplificateur ou simplement une coïncidence que certains cas ont été détectés sur et autour de ce marché », a déclaré Peter Ben Embarek, expert de l’OMS en matière de sécurité alimentaire et de zoonoses, lors d’une conférence de presse virtuelle à Genève au début du mois. » (source CNEWS)
Dans ce brouillamini de désinformation qui dessert la Chine, émerge alors la possibilité décrite par Colin Carlson, professeur à l’université de Georgetown, USA, et spécialiste de la propagation des virus zoonotiques, qui évoque l’hypothèse de la présence sur le marché de Wuhan d’un « super propagateur » au début de l’épidémie. C’est-à-dire un individu qui aurait transmis le virus à beaucoup plus que les 2 à 3 personnes que contamine en moyenne un malade du coronavirus. Les super contaminateurs existent bel et bien, comme ce médecin de Canton qui avait soigné des patients du SARS et qui avait lui-même fini par contaminer en quelques minutes, le 21 février 2003, 16 personnes qu’il avait croisées dans le couloir de l’hôtel Hilton de Hong Kong avant de mourir dans les jours qui suivaient. De même l’épidémie de Covid-19 a éclaté en Alsace probablement suite à un rassemblement religieux de 2000 évangélistes organisé par l’église de la Porte Ouverte Chrétienne dans le quartier de Bourtzwiller à Mulhouse. On ne sait exactement qui était présent à ce rassemblement assez confidentiel, avec des participants venus potentiellement de toute la France et même de l’étranger, mais on pense qu’un seul malade aurait pu en contaminer quelques dizaines en un week-end, sans compter un nombre important de Chinois présents dans la région (Colmar) en raison d’émission de TV réalité sur la cuisine, à destination de la Chine. Finalement ce n’est pas un hasard si l’épidémie de SARS-Cov2 a véritablement explosé en France à partir de la conurbation Colmar-Mulhouse dans le Haut-Rhin. Mulhouse est jumelée avec Bergame qui est la ville martyre italienne la plus effroyablement touchée par le Covid-19 et d’où est partie l’épidémie en Italie le 25 janvier 2020. Avec son aéroport international à bas coût (Bergamo Orio al Serio), Bergame était avant l’épidémie le lieu de transit d’innombrables travailleurs chinois, dont une partie venait de Wuhan, et qui étaient employés dans le textile et la confection dans la région autour de Milan. Jining, qui a été le deuxième centre épidémique en Chine après Wuhan, était également jumelée avec Mulhouse.
Tout cela masque la réalité de la situation et il faut rester très prudent vis-à-vis de toutes les déclarations officielles provenant de la Chine, de l’OMS ou de nos propres gouvernants qui jusqu’à maintenant n’ont pas fait preuve d’un grand discernement dans la prévention et la gestion de l’épidémie.
En résumé, la Chine n’a pas mené d’enquête officiellement auprès des tout premiers patients ayant présenté des symptômes respiratoires dus à une pneumonie atypique (chapitre 1) en dehors du marché du Huanan entre la mi-novembre et début décembre. Elle se contente de dire : (1) que le virus ne vient pas initialement du marché de Huanan et (2) laisse le soin aux médias occidentaux d’affirmer que le virus est d’origine animale. Une recette qui l’arrange au plus haut point.
En ce qui concerne les marchés aux animaux sauvages, suite à l’épidémie mortelle de SARS de 2003 qui a été contenue à temps, le gouvernement chinois ne les a pas fermés malgré la demande pressante de l’OMS et d’autres instances internationales. Elle n’a toujours pas l’intention réelle de les fermer. Comme d’habitude le flou le plus artistique règne entre la réglementation chinoise d’interdiction de trafic et de vente d’animaux sauvage, instaurée après 2003, et la réalité sur le terrain. La santé de l’humanité est prise en otage par la Chine. L’analyse brute des chiffres montre qu’avec seulement 4600 morts et une épidémie maîtrisée la Chine vient de remporter par la force des évènements une victoire écrasante contre l’occident et surtout sur son frère ennemi que sont les USA où on dénombre, au 16 septembre 2020, plus de 200.000 morts du Covid-19. Aujourd’hui, 6 octobre 2020, 9 mois après sa révélation au monde la pandémie court encore un million de morts au total dans le monde. Mais ce chiffre est dérisoire comparé à ce qui pourrait arriver dans le futur si la Chine s’obstinait à maintenir une triple menace sur le monde avec ses marchés aux animaux sauvages, ses recherches universitaires civiles sur les coronavirus GOF et ses recherches militaires plus ou moins affichées.
À la différence du SARS-Cov de 2003, il plane un énorme doute sur l’origine naturelle du virus de SARS-Cov2 de 2019
L’origine du virus n’est pas établie de façon certaine et son génome présente des caractéristiques très troublantes pour lesquelles une explication naturelle semble peu vraisemblable selon un bon nombre de chercheurs qui n’hésitent pas à l’affirmer publiquement, surtout à l’étranger. Pas mal d’autres se contentent de l’affirmer dans leur cercle amical, sans oser toutefois en témoigner ouvertement tant il est à présent difficile, et surtout dangereux professionnellement, de porter un avis critique en France sur quoi que ce soit qui ne rentre pas dans la pensée consensuelle. En tant qu’auteur de ce texte nous pensons que cette pensée unique est imposée par les médias au service de l’oligarchie mondialiste, mais nous admettons qu’il s’agit là d’un avis purement politique. Laissons le lecteur interpréter cet état de fait de la façon la plus indolore qu’il souhaite pour la représentation de la société dans laquelle nous vivons.
Comme nous l’avons vu au chapitre 1, il y a eu destruction de preuve, rétention d’information et défaut volontaire d’investigation scientifique de la part de la Chine. De ce fait la question de l’hôte intermédiaire, qui serait le pangolin, est devenue très complexe, car elle n’a pas pu être résolue avec suffisamment de fiabilité et soulève beaucoup d’interrogations comme nous le verrons dans le prochain chapitre. Cela conforte évidemment la thèse non officielle à caractère complotiste que le virus ne proviendrait pas du marché du Huanan, mais plutôt de l’institut de virologie de Wuhan et de son mythique laboratoire P4 dont l’institut est doté depuis 2015.
La réalité semble avoir dépassé la fiction avec l’interview spectaculaire du Pr Montagnier, prix Nobel de Médecine pour la découverte du virus du Sida, diffusée le 17 avril 2020, sur une chaîne de télévision grand public. Dans cette longue interview, il affirme que le génome du virus SARS-Cov2, responsable de la pandémie, présente des éléments exogènes d’information (EEI) qui ne peuvent pas être d’origine naturelle. Nous consacrons l’avant-dernier chapitre de cet ouvrage aux travaux du Pr Luc Montagnier et du mathématicien Jean-Claude Perez qui leur permettent de tirer cette conclusion terrible. Soulignons qu’ils ne sont pas les seuls et que de nombreuses interrogations ont été soulevées par d’autres chercheurs scientifiques établis, concernant le caractère naturel peu vraisemblable de certaines parties de la séquence génomique du virus, en particulier la présence d’une région de la séquence qui n’est apparentée à aucun autre coronavirus connu.
La thèse officielle, défendue par les chercheurs de l’Institut Pasteur, qui font référence en France en la matière, est que cette région qui semble provenir de nulle part est le fruit d’une recombinaison naturelle entre virus opérée par la nature. Dans une forme de raisonnement elliptique, cher à la communauté de recherche française et que l’on retrouve dans de nombreuses thèses, leur conclusion est que de toute évidence la nature est capable de faire ce type de réarrangement puisqu’il est observé, en excluant toute autre possibilité… Nous verrons cependant que ce raisonnement imparable se heurte pourtant à la théorie des probabilités et au fait que de nouvelles méthodes de génie génétique permettent depuis quelques années d’éditer un génome et d’y insérer des éléments de séquence, voire des gènes complets, sans laisser de trace de manipulation visible.
Partie 4 — Les virus militaires de Zhoushan
La recherche académique militaire semi-confidentielle est très développée en Chine
Nous relatons ici une recherche conduite sur les coronavirus de chauves-souris, entre 2015 et 2017, au sein de l’Institut de Recherche Médicale de l’Hôpital Général de l’Armée de Libération Populaire de la région de Nanjing où se situe le commandement de la région militaire de la Chine de l’Est. Parmi les 15 auteurs de cette recherche, 11 sont affiliés à ce grand centre de recherche hospitalier militaire. Le premier et le dernier auteur (Dan Hu et Changjun Wang) sont également affiliés à La Troisième Université Militaire chinoise, fondée en 1954, qui se situe à Chongquing. Nous ne parlons pas ici de recherches secrètes, mais nous voyons comment l’autorité militaire chinoise abrite intelligemment des recherches universitaires semi-civiles dans ce domaine. Cela leur permet d’avoir des chercheurs de grande qualité développant des outils techniques et des connaissances qui peuvent alimenter par la suite des programmes ultra-secrets, bien qu’en l’occurrence nous ne disposons d’aucune preuve, mais seulement d’indices circonstanciels de ce qui pourrait potentiellement se passer ou pas dans ce domaine.
De façon amusante pour les agents de renseignements, on peut noter que parmi les 15 auteurs, tous chinois, de cette étude publiée, se trouve un auteur non chinois affilié à l’Université de Stony Brook aux USA ! On pourrait y voir là une tentative peu discrète d’infiltration par les USA des recherches militaires universitaires chinoises sur les coronavirus de chauves-souris. En tout cas, c’est bien par les collaborations universitaires que l’on obtient les informations les plus sûres en matière de développement technologique. Bien sûr, en acceptant un chercheur étranger les autorités chinoises savaient ce qu’elles faisaient… Pour tout ce qui concerne espionnage, contre-espionnage, manipulation et contre-manipulation, nous renvoyons le lecteur au chapitre consacré à ce sujet dans l’excellent ouvrage de Sun Tsu (L’Art de la Guerre, paru vers 500 av. J.-C).
Dan Hu relate que pour mieux comprendre la relation évolutionnaire entre les virus SARS-Cov et leurs réservoirs, 334 chauves-souris recouvrant plusieurs espèces dont une des deux dites en fer-à-cheval (Rhinolophus ferrumequinum) ont été capturées en 4 occasions, entre 2015 et 2017, dans des grottes montagneuses aux environs de la cité de Zhoushan dans la province du Zhejiang. Ces chauves-souris ne présentaient aucun signe de maladie au moment de leur capture.
Les génomes complets de deux coronavirus extraits ont été publiés : « L’analyse complète de deux nouveaux SL-CoVs de Zhoushan (ZXC21 et ZC45) montre que leurs génomes avaient une longueur respective de 29.732 nucléotides (nt) et 29.802 nt avec 13 cadres de lecture ouverts (open reading frames ou ORF en anglais) [NDA : il s’agit de parties de la séquence de nucléotides qui forment le code génétique actif du virus, délimitées par des codons d’initiation et d’arrêt. Les ORF peuvent être transcrites par des enzymes spécialisées en séquences d’acides aminés formant les protéines qui permettent au virus d’exister et de fonctionner]. Ces deux nouveaux virus partagent seulement 81 % d’identité de séquence génomique avec les virus SARS CoVs isolés chez l’homme et la civette en 2003. De ce point de vue, ils sont donc plus distants du SARS-Cov que la plupart des autres SL-CoVs de chauve-souris, identifié en Chine depuis 2003, qui se trouvaient dans la zone des 90 % d’identité de séquence.

Les écrits de Dan Hu et Changjun Wang nous aident à ouvrir les yeux sur la possibilité réelle d’un programme militaire
Nous pouvons remercier Dan Hu et Changjun Wan, qui ont endossé la responsabilité de superviser les recherches publiées, pour leur transparence. Malgré le fait qu’ils travaillent dans des institutions militaires sous contrôle de l’armée rouge ,ils bénéficient d’une large marge de manœuvre en ce qui concerne la publication de leurs travaux. Comme nous l’avons expliqué précédemment les militaires sont dépourvus de génie scientifique et leur stratégie consiste donc à attirer d’excellents chercheurs universitaires pour travailler dans des laboratoires plus ou moins confidentiels. Cela n’est possible qu’en leur laissant une certaine latitude dans la communication avec leurs pairs sur le plan international. Un chercheur scientifique qu’on empêche de communiquer se sentira frustré et isolé et finira par ne plus rien produire et s’en aller. D’où l’intérêt de les laisser publier. Ils se sentent reconnus et valorisés ce qui est tout à fait louable. De plus cela permet d’organiser des échanges académiques internationaux qui peuvent s’avérer fructueux.
Leurs travaux nous permettent d’obtenir beaucoup de renseignements pour déduire l’ampleur du répertoire de virus que possède potentiellement l’armée chinoise ! Voici, en substance, ce qu’ils rapportent dans la première de leurs deux publications phares de 2017 et 2018.
Ils ont analysé les types de virus pathogènes présents chez les chauves-souris de la zone côtière sud-est de la Chine, en procédant au séquençage méta-génomique par PCR (réaction de polymérisation en chaîne) d’animaux provenant de six sites sentinelles de ces zones côtières. L’acronyme PCR est à présent assez bien connu des Français puisque les tests de positivité au virus SARS-Cov-2 sont réalisés par cette technique qui permet d’amplifier en les multipliant par milliards les fragments d’ADN ou d’ARN présents sur un prélèvement nasal ou bien sur un tampon frotté sur une surface.
En utilisant la technique PCR, ils ont obtenu des millions de fragments de séquences d’ARN viral [NDA, les fragments de séquences ont une longueur d’environ 100 nt, c’est-à-dire environ 1/250 de la longueur totale d’une séquence de coronavirus] provenant des intestins et des tissus pulmonaires de 235 chauves-souris. Ils ont ensuite produit un travail colossal pour déchiffrer le code génétique de tous les fragments de séquences amplifiés en utilisant très probablement des batteries de robots de séquençage. Une fois le code des fragments de séquences établi, ces codes sont analysés par des logiciels spécialisés qui permettent d’identifier, par comparaison avec des banques de données internationales, à quel type d’organisme (bactéries, virus, plante, vertébrés…) et quel type de gène ces fragments de séquence peuvent appartenir. C’est un travail long et rigoureux que l’on peut cependant programmer assez facilement sur des ordinateurs puissants. De proche en proche, par comparaisons multiples et superpositions partielles on peut réassembler l’ensemble des séquences fragmentées jusqu’à obtenir en principe les séquences complètes des organismes auxquels elles appartiennent.
Même informatisé, ce travail prend énormément de temps, car il nécessite souvent l’intervention humaine pour pallier aux difficultés que rencontrent les logiciels et ne peut être conduit avec succès à l’heure actuelle que sur des génomes de virus, car ils sont extrêmement courts. Cependant, plus le génome que l’on cherche à établir sera éloigné d’une séquence de virus déjà connu et plus sa reconstitution posera problème.
Les séquences partielles de virus (viromes) leur ont permis d’identifier 25 familles virales, incluant 16 types distincts de virus d’organismes vertébrés, quatre de virus de plantes et cinq d’insectes. Tous les échantillons ont été analysés phylogénétiquement et de nombreux nouveaux virus de chauves-souris ont été découverts. Certains sont très proches de pathogènes humains et animaux connus, incluant : coronavirus, norovirus, adénovirus, bocavirus, astrovirus, et circovirus.
En ce qui concerne les coronavirus de type SARS, leur présence dans un fouillis de milliards de séquences partielles est repérable grâce aux protéines non structurales RdRp codées dans l’ORF1 qui sont très conservées et spécifiques à ces virus. Les enzymes RdRp (ARN réplicases) catalysent la synthèse d’un brin d’ARN complémentaire à partir d’un brin d’ARN servant de matrice. Elles sont indispensables pour la réplication des virus à ARN comme les coronavirus. Sans avoir besoin d’établir la séquence complète de tous les virus identifiés, on peut par l’intermédiaire d’un fragment spécifique de ces protéines établir un arbre phylogénique décrivant la distance évolutive entre les séquences des nouveaux virus identifiés (degré de parenté entre elles). De cette façon, ils ont pu identifier 62 alpha-coronavirus et 335 beta-coronavirus…
Selon leurs écrits, ils ont établi les séquences complètes de 9 nouveaux beta-coronavirus et 7 nouveaux alpha-coronavirus dont ils ont établi le degré de parenté (arbre phylogénique) entre eux ainsi qu’avec d’autres représentants connus de ces familles de coronavirus.
Leurs observations démontrent la très grande variété de ces virus, chaque colonie de chauves-souris étant le réservoir évolutif de centaines de ces virus pendant un temps que les auteurs estiment long. C’est un euphémisme très spécial ! L’analyse phylogénique du nombre très restreint de coronavirus de chauves-souris identifiés permet d’estimer la présence d’un ancêtre commun il y a 10.000 ans. Évidemment plus on ajoutera de séquences à l’arbre phylogénique et plus le ou les ancêtres communs s’éloigneront dans le passé. La réalité c’est que ces virus sont là très probablement depuis bien plus longtemps, probablement des millions d’années.
Dan Hu poursuit en soulignant qu’ils ont effectué des tests de pathogénie sur ces deux virus, démontrant qu’ils pouvaient se répliquer [NDA, ce qui leur permet de vérifier et d’affirmer que leur séquence est bien reconstruite correctement] et causer une maladie respiratoire chez les rats de laboratoire. Ils ont même observé au microscope électronique que des particules virales se retrouvaient dans le cerveau des rats… On voit donc que l’annonce faite à retardement que le SARS-Cov2 pouvait causer des troubles neurologiques tels que des troubles olfactifs (qui sont ensuite devenus le symptôme avant-coureur le plus courant) n’est qu’une pseudo-découverte car certaines personnes savaient très bien que cela pouvait être le cas dès le départ de l’épidémie.
Ils concluent que leur étude élargit la compréhension des chauves-souris en tant que réservoir viral et fournit une base pour l’étude de la transmission des virus de la chauve-souris à l’être humain. Une base de connaissances certes, mais également une base de matériel génétique à l’instar des travaux de Shi Zheng Li et Ralf Baric sur les virus synthétiques GOF.
Au total combien y avait-il de nouvelles séquences génomiques parmi les centaines de coronavirus identifiés ? Ont-elles toutes été reconstituées et déposées dans les banques de données scientifiques internationales ? Nul ne le sait.
Quelle peut-être la vision des stratèges militaires chinoises ?
La Chine en tant qu’empire s’est développée depuis plus de deux mille ans, après une phase de guerres incessantes entre les Royaumes combattants. Cette période s’est étalée entre le 5e et le 2e siècle av. J.-C. Selon le général Sun Tzu, l’auteur du premier ouvrage de stratégie militaire universellement connu qui date de cette époque : « la guerre est pour l’état une préoccupation sérieuse ; elle nécessite une étude approfondie. » Nous n’écrivons pas cela pour signifier que les Chinois sont belliqueux de nature, mais au contraire pour dire que ce sont des gens très avisés ayant une tradition et une compréhension militaire de culture très ancienne.
Jusqu’à présent les armes bactériologiques étaient considérées comme étant impossibles à mettre en œuvre sur un théâtre d’opérations sans risquer d’y succomber à son tour. La fabrication et le stockage de bactéries et de virus hautement pathogènes sont également très délicats et peu pratiques sur le plan de la gestion et la mise en œuvre militaire. Cela était suffisamment gênant, durant la guerre froide, pour que les grandes puissances renoncent au développement opérationnel de ce genre d’armes au profit de l’arme nucléaire qui semblait bien plus terrifiante et contrôlable à l’époque. Elle le serait d’ailleurs certainement toujours sans la réalité émergente des virus synthétiques GOF aux pouvoirs contagieux et pathogènes renforcés en même temps. Sans cela, le concept de virus exterminateur serait à relativiser, car en principe il y a toujours une certaine proportion de la population qui ne contracte pas le virus ou reste totalement asymptomatique.
Toutes les maladies les plus terribles ont par le passé présenté une limite de létalité. La peste noire (l’agent pathogène est le bacille Yersiniae Pestis) de 1720 n’a tué que la moitié des habitants de Marseille dont il était impossible de sortir, un cordon militaire ayant été établi autour de la ville. Au niveau de la Provence entière, elle aussi séparée du reste de la France par le mur de la peste, la maladie n’a tué que 30 % de la population. Même les fièvres hémorragiques de type Ebola ou Marburg, qui sont les maladies contagieuses les plus létales, l’intégralité des populations touchées ne meurent pas. Le degré de létalité varie entre 40 et 80 % selon les souches virales.
Cependant, comme on le voit avec le SARS-Cov2, toutes ces maladies ne laissent pas forcément les survivants indemnes de toutes séquelles. Dans le cas d’Ebola, les survivants ayant développé des symptômes présentent des lésions d’organes qui restent potentiellement invalidantes à vie.
Fort heureusement, le mode de contagion des virus les plus létaux comme celui de la rage ou du Sida nécessite un contact ultra rapproché avec leur vecteur (morsure, contact de salive avec une coupure, rapport sexuel). On ne sait pas exactement comment le virus du Sida est passé du singe à la l’homme, mais il est létal à 100 %. La trithérapie est un traitement lourd qui permet de reculer considérablement l’échéance, mais pas de guérir. Le Sida tue 700.000 personnes chaque année. Celui de la rage est également létal à 100 % une fois les symptômes déclarés. Une équipe médicale a réussi l’exploit de sauver une adolescente qui avait été mordue environ 3 semaines auparavant et présentait les symptômes de la rage. Le virus de la rage pénètre le système nerveux central et la douleur engendrée ainsi que les spasmes cardio-vasculaires finissent par tuer la personne atteinte.
Lorsqu’elle est arrivée à l’hôpital, cette jeune fille ne pouvait plus parler et était quasi inconsciente. Pour la sauver, elle a été mise en coma artificiel pendant une semaine, les fonctions du cerveau étant suspendues le temps que son système immunitaire puisse construire une réponse au virus. Elle a reçu également deux antiviraux puissants, ribavarine et amantadine. Sa convalescence sous traitement de vitamine B a duré 6 mois et il a fallu attendre 1 an avant qu’elle ne recouvre l’intégralité de ses facultés ! Nous voyons bien que si un tel virus devenait contagieux au-delà du mode de transmission directe par morsure ou léchage, alors l’humanité serait réellement en danger.
Bien sûr, comme nous l’avons montré dans ce chapitre et les précédents des laboratoires secrets ont existé dans le passé, de façon certaine aux USA et en Russie et très probablement également en Chine. Aujourd’hui, l’armée chinoise dispose, en plus de milliers de colonies de chauves-souris, de toutes les ressources humaines dans les domaines de la microbiologie et de la virologie en plus de la technique de construction de laboratoires de haute sécurité. Elle contrôle également des campus militaires où la recherche de haut niveau dans le domaine de la synthèse de virus GOF pathogéniques mortels, potentiellement pandémiques, peut être conduite discrètement sans la nécessité de créer une zone ultra-secrète qui attirerait l’attention par le bouche-à — oreille. Savoir si des laboratoires totalement secrets existent est donc anecdotique.
En tout cas, les laboratoires secrets posent de nombreux problèmes de sécurité et les militaires ne sont jamais très performants dans la conduite de recherches scientifiques. Les militaires chinois savent qu’ils doivent laisser cela aux universitaires. Leur stratégie dans ce domaine est probablement la même que celle des USA. Il s’agit visiblement de laisser la recherche universitaire s’opérer librement afin de disposer de toutes les pièces du puzzle prêtes pour ensuite être assemblées ultra rapidement en cas de besoin. Mais y en aura-t-il jamais besoin quand on dispose par ailleurs d’une armée conventionnelle surpuissante et de l’arme thermonucléaire comme moyen de dissuasion ? L’utilisation de l’arme virale relèverait de la folie pure et simple.
Les virus ne peuvent se concevoir donc que comme une arme de terrorisme qui ne dirait pas son nom sous couvert d’épidémie naturelle. Comme nous le verrons dans un prochain chapitre, il est à l’heure actuelle possible de créer des virus synthétiques sans que leur génome ne porte les stigmates des manipulations effectuées. La seule façon de les identifier réside dans des éléments subjectifs ne pouvant apporter de preuve absolue comme une phylogénie mal établie, l’absence d’un hôte intermédiaire identifié, l’absence de mutations d’adaptations en début d’épidémie et un défaut d’harmonie du génome qui finirait par le rendre rapidement instable.
Une chose est certaine, la pandémie de Covid-19 démontre la difficulté immense qu’il y aurait à contrôler la dévastation qu’une telle arme pourrait répandre sur terre si un virus 10 à 100 fois plus létal s’y répandait. Rappelons que la grippe espagnole a été environ 150 fois plus mortelle (en proportion de la population terrestre) que ne l’a été le COVID-19 pour l’instant avec 1 million de morts entre décembre 2019 et septembre 2020. Que se passerait-il ? La Chine vient de démontrer qu’elle a pu contrôler une pandémie sur son sol, par des mesures drastiques, en fermant totalement la ville de Wuhan, ainsi que la ville de Pékin pour la mettre à l’abri, en plus d’un confinement domestique inhumain. Les pays de l’UE, manipulés par la Chine et l’OMS n’ont pas su fermer leurs frontières à temps et pratiquer des tests et des mises en quarantaines systématiques à l’entrée de leurs territoires. Il s’agissait là d’une absence de réponse plus idéologique que raisonnée. Le grand credo de nos gouvernants européistes était de préserver le libre-échange, quels que soient les évènements. Gageons que ce credo insensé ne perdurera pas à la prochaine pandémie de coronavirus qui ne manquera pas de se produire dans les années à venir si la Chine ne prend aucune mesure radicale concernant les marchés aux animaux sauvages, les trafics de chauves-souris en particulier, et les recherches sur les virus GOF pandémiques.
chapitre 5 — Les propriétés d’infectiosité et de réplication hors norme du SARS-Cov2 sont-elles à l’origine de la paranoïa de nos élites gouvernementales ?
À l’heure où un second confinement qui sera pénible et dévastateur pour l’économie de la France commence, on peut se demander si une paranoïa n’a pas gagné nos gouvernants en ce qui concerne la possible résilience pandémique au long court du SARS-Cov2. Au cœur du problème se trouve une capacité extraordinaire du virus à réparer son génome et à infecter les cellules.

Partie 1 — Fonctionnement des coronavirus à SARS de 2002 et 2019
Comme le SARS-Cov de 2002-2003 (à présent appelé également SARS-Cov1), le SARS-Cov2 apparu fin 2019 est un virus potentiellement très résilient grâce à une propriété d’auto-réparation de son génome. C’est une caractéristique exceptionnelle absolument unique chez les virus qui permet à son extraordinaire ARN polymérase, d’être dix fois plus rapide que celle des autres virus. À cela s’ajoute une capacité accrue à reconnaître le récepteur de pénétration cellulaire ACE2 par l’intermédiaire d’une protéine de l’enveloppe virale (hémagglutinine estérase) abusivement appelée « seconde clé » de façon journalistique anxiogène. En fait, cette protéine non essentielle en apparence permet au virus de mieux adhérer aux cellules hôtes augmentant les chances de la protéine S de reconnaître le récepteur de pénétration cellulaire. Cette aide à la pénétration cellulaire n’est pas unique au SARS-Cov2, on la retrouve dans de nombreux virus super infectieux comme le virus Ebola, le VIH et les souches hautement pathogènes de la grippe aviaire.
De façon générale, les betacoronavirus, dont font partie le SARS-Cov1 (2002) et le SARS-Cov2 (2019), sont des virus dits à ARN non segmenté de polarité positive. Ces termes de jargon scientifique sont peu parlants et a priori rébarbatifs pour le public, mais ils ont une grande importance, car ils correspondent à des caractéristiques essentielles qui conditionnent le mode de réplication de ces virus et nous éclairent sur ce qui peut inquiéter dans les milieux informés (dont font partie nos gouvernants).
Sur le plan de l’assemblage moléculaire
Tous les coronavirus possèdent une double enveloppe protéique encapsulant du matériel génétique sous forme d’ARN (acide ribo-nucléique) dit ARN au lieu d’ADN (acide désoxyribonucléique) classique qui constitue le code génétique de l’intégralité des espèces du monde vivant et également d’un certain nombre de virus. Dans le cas des coronavirus cet ARN se présente sous la forme d’un seul brin non segmenté. La polarité + de ce brin signifie qu’il va être directement transcrit en protéines par l’intermédiaire des ribosomes (organites cellulaires) comme de l’ARN messager classique. Dans certains autres types de virus, l’ARN est de polarité — ce qui signifie qu’il doit être transcrit en ARN + (par une transcriptase spécialisée) avant d’être transcrit en protéines.
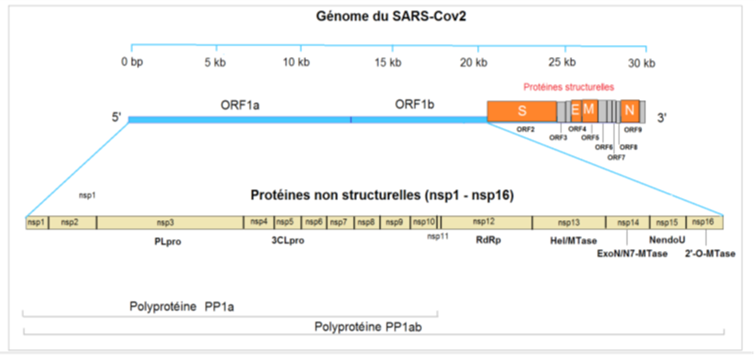
Les protéines structurales
Trois protéines virales sont ancrées dans l’enveloppe virale : la protéine spicule (S), qui lui donne son aspect en couronne et son nom, la protéine de membrane (M), la protéine d’enveloppe (E).
Les protéines M et E sont impliquées dans l’assemblage viral et la sécrétion. La protéine S s’assemble en trimères à la surface des virions et joue un rôle clé dans l’entrée du virus dans la cellule cible. Elle est constituée de deux domaines, le domaine S1 responsable de la liaison du virus à son récepteur et le domaine S2 responsable de la fusion de l’enveloppe virale avec une membrane cellulaire.
La fusion est activée par des protéases cellulaires par clivage de la protéine S. La protéine de membrane M est la plus abondante de l’enveloppe et joue un rôle majeur dans l’assemblage du virion. La petite protéine d’enveloppe E a un rôle dans la sécrétion des virions.

La capside virale est formée par la nucléoproteine (N) qui encapsule le génome viral. Le coronavirus SARS-CoV-2 utilise comme porte d’entrée dans les cellules qu’il infecte une molécule présente à leur surface : l’enzyme transmembranaire ACE2 qui se comporte comme un récepteur pour le virus. L’entrée du SARS-CoV-2 dans les cellules cibles nécessite l’activation de la protéine S présente à la surface du virus.
Finalement l’hémagglutinine estérase (HE), une protéine de surface, est impliquée dans le mécanisme de pénétration cellulaire en permettant l’adhérence du virus sur la surface des cellules hôtes.
Les récepteurs ACE2 sont présents sur les cellules qui tapissent l’intérieur des alvéoles pulmonaires également sur l’intestin grêle et à un degré moindre sur le gros intestin et le foie.
Les protéines non-structurales (réplicases)
Les protéines non-structurales codées par les cadres de lecture ouverts (ORF1a et ORF1b) sont au nombre de 16 (nsp1 à nsp16). Elles participent à la réplication du génome du virus et la régulation de ce processus, mais également potentiellement à un mécanisme d’évasion immunitaire (nsp3, nsp13, nsp15, nsp16).
Les facteurs accessoires ou auxiliaires
En plus des gènes classiquement conservés à travers les coronavirus, la présence de 6 gènes additionnels a été identifiée à l’extrémité 3′ du SARS-Cov1 de 2002 (ORF 3a, 3 b, 6 7a, 7 b, 8a, 8 b et 9 b). Ce sont des protéines accessoires (facteurs accessoires ou dits auxiliaires) dont les fonctions distinctes précises restent très mal connues, mais sont globalement impliquées dans l’abaissement ou la suppression de la réponse immunitaire.
Le SARS-Cov2 encode une série identique de facteurs accessoires supplémentaires 3a, 3b, 6, 7 a, 7 b, 8, 9 b, très semblable au SARS-Cov1 avec un degré d’identité troublant pour certains facteurs.
La protéine codée par l’ORF3a participerait au blocage de la signalisation des interférons, via la régulation négative de l’expression d’IFNAR (rôle de la protéine ORF3a du SARS-CoV-1) et de la phosphorylation de STAT-1 (par la protéine nsp3).
L’ORF3b et l’ORF6 sont des antagonistes démontrés de l’INF-1.
Le facteur codé par l’ORF7a, une petite protéine composée de 122 acides aminés, partage 100 % d’identité avec celui du SARS-Cov1. La fonction de cette protéine de type transmembranaire reste totalement inconnue en raison du fait que malgré un type de repliement commun à 180 autres protéines connues sa composition en acide aminé est si différente que sa fonction ne peut être inférée par comparaison.
Le rôle de l’ORF7b est jusqu’ici inconnu, probablement à la fois auxiliaire et structural.
La protéine de SARS-CoV2 codée par l’ORF8 montre une homologie de séquence de moins de 20 % par rapport à celle de l’ORF8 trouvé sur le SARS-CoV-1 et est donc hautement divergente. Il a été proposé que l’ORF8 pouvait nuire à la fonction immunitaire de l’hôte de plusieurs façons. Les études ont montré que l’ORF8 du SAR-CoV-2 (mais pas celui du SARS-CoV-1), entraîne une suppression des complexes majeurs d’histocompatibilité (MHC-1) un élément essentiel de la réponse immunitaire. La sur-expression exogène de l’ORF8 perturbe également la signalisation de l’interféron 1 (IFN-1) un médiateur de la réponse immunitaire.
La protéine de l’ORF9b (sous unité du gène nucléocapside ORF9) est un suppresseur de l’interféron-1 par l’intermédiaire de la formation d’un complexe avec la protéine TM70 intermédiaire de signalisation indispensable dans la réaction immunitaire innée.
Nous voyons donc que le SARS-Cov2 par l’organisation de son génome, l’assemblage de sa structure tridimensionnelle, et la présence d’une série quasi identique de facteurs auxiliaires situés à l’extrémité 3′ de son ARN est extrêmement semblable au SARS-Cov1 avec lequel il partage 82 % d’identité (ndlr : clarification de l’auteur) de séquence en ce qui concerne les nucléotides codant les gènes, c’est-à-dire l’ensemble des protéines que nous venons de décrire. Cependant, si on inclut les parties non codantes du génome le pourcentage d’identité tombe à 79 %. Ce relativement faible pourcentage a été un prétexte en début d’épidémie à l’affirmation répandue que ce virus était différent. Mais il s’agit d’une distance évolutive sur le plan phylogénique et non pas sur le plan de la structure et du fonctionnement du SARS-Cov2, comme nous venons de le voir. De plus, il partage avec le SARS-Cov1 une capacité de réparation de son ARN unique parmi les coronavirus ce qui le rend encore plus proche de celui-ci.
Sur le plan du génome et du mode de réplication du SARS-Cov2
De façon générale, sur le plan moléculaire, l’ARN est très proche de l’ADN dont il est une copie sur une courte longueur. Il correspond à quelques gènes seulement, souvent uniquement un, correspondant à la synthèse d’une ou plusieurs protéines. Dans le fonctionnement des cellules, il est l’intermédiaire moléculaire indispensable qui permet l’expression des gènes. Dans toutes les cellules eucaryotes (c’est-à-dire l’essentiel du monde vivant), l’ARN est généré dans le noyau sous forme de simples brins au lieu de double hélice comme l’ADN. Cependant, dans certaines circonstances il peut aussi former aussi de doubles hélices. Du fait qu’il est exprimé sous forme de simple brin et qu’il n’est pas à proprement parlé le vrai code génétique, il n’y a pas de mécanisme cellulaire de réparation de l’ARN, la réparation des gènes se faisant par l’intermédiaire d’enzymes spécialisées directement au niveau de l’ADN. Sa réparation s’effectue grâce au fait qu’il est sous forme de double hélice. Les 2 brins de la double hélice d’ADN étant complémentaires l’un de l’autre, si l’un des 2 brins seulement est endommagé, c’est-à-dire lorsqu’un des nucléotides composant un filament (brin) moléculaire d’ADN est manquant (cassure) ou ne correspond pas aux nucléotides complémentaires, ou est corrompu chimiquement, alors des enzymes spécialisées entrent en jeu pour réparer le brin endommagé en se servant de l’autre brin comme référence pour la réparation. Des mécanismes sont également prévus pour la rupture double brin, fort heureusement plus rare, car beaucoup plus dangereuse pour la survie saine de la cellule.
À noter que les endommagements ou ruptures de brin peuvent être causés par les enzymes responsables de la traduction et de la réplication des gènes, mais avec une probabilité plus faible cependant que ceux causés par le métabolisme cellulaire normal produisant du stress d’oxydation, source de nombreux dommages. Les rayonnements ultraviolets (UV) et les radiations ionisantes ou les agents chimiques génotoxiques sont également la source de rupture simple ou double de l’ADN. Ces dernières étant les plus dangereuses, car pouvant déclencher une réplication anormale des cellules qui ont perdu une partie de leur génome (cassure double brin non réparée) ou ont muté (cassure double brin mal réparée).

Du fait que l’ARN est produit et circule majoritairement sous forme simple brin non réparable, s’ajoute une instabilité chimique intrinsèque due à ce qu’une des briques essentielles de sa structure moléculaire est la molécule de ribose moins stable que le désoxyribose. Par conséquent, et contrairement au virus à ADN, les virus à ARN présentent des taux de mutation élevés au cours de leur réplication, car la machinerie cellulaire ne prévoit pas leur réparation comme c’est le cas pour l’ADN.
Ces nombreuses mutations engendrent des variations dans leurs niveaux de contagiosité et de virulence qui sont observées entre des épidémies distinctes et également durant une même épidémie, comme c’est le cas du virus SARS-Cov2 causant la Covid-19. Comme nous le verrons dans le chapitre suivant, cette caractéristique particulière permet de tirer des conclusions intrigantes à propos du SARS-Cov2 par rapport au SARS-Cov1 de 2002-2003 qui remettent sérieusement en question le fait que le pangolin puisse être l’hôte intermédiaire et indiquent une adaptation très précoce du virus à l’homme.
C’est une particularité essentielle des virus à ARN de pouvoir se répliquer en se passant de code ADN. Cela tient au fait que les virions ne se répliquent pas eux-mêmes, mais utilisent une partie de la machinerie cellulaire de leur hôte pour se reproduire.
L’ARN est en fait un élément intermédiaire dans l’expression d’un gène, entre l’ADN formant les chromosomes contenus dans le noyau de la cellule et la synthèse protéique se produisant en dehors du noyau dans le cytoplasme. Cette synthèse nécessite spécificité et production de masse. Dans le processus d’expression des gènes encodés par l’ADN (stable sous forme de double hélice), de l’ARN est synthétisé par une enzyme (l’ARN polymérase) sous forme de multiples brins identiques codant la même protéine ce qui permet leur production en grand nombre par les ribosomes, sorte de petites usines de fabrication, situés dans le cytoplasme.
Les coronavirus court-circuitent donc toute la phase d’expression génétique du noyau cellulaire en infestant directement le cytoplasme d’une cellule hôte dans lequel ils relarguent leur ARN, comme si c’était de l’ARN messager propre à la cellule. Il détourne les ribosomes pour fabriquer les protéines indispensables à la réplication virale et son encapsulation. Parmi ces protéines, une enzyme appelée ARN réplicase (ou ARN polymérase – ARN dépendante, RpRd) indispensable à la réplication de l’ARN viral est générée par les ribosomes. Le matériel génétique du virus code donc une protéine qui une fois exprimée dans la cellule hôte va permettre de répliquer son ARN.
Dans le détail le mécanisme de réplication d’un coronavirus peut se résumer ainsi : « Le génome de polarité positive est traduit directement en une grande polyprotéine qui est ensuite clivée pour donner naissance à trois protéines P1, P2 et P3. La maturation de ces protéines virales fait intervenir plusieurs clivages en cascade. La région P1 contient l’information génétique codant pour les protéines de capside. Les régions P2 et P3 codent pour des protéines non structurales, dont l’ARN polymérase-ARN dépendante ou réplicase. La réplicase va synthétiser un brin (— ) pour aboutir à la forme réplicative constituée d’un brin (—) et d’un brin (+) appariés en double hélice. Le brin (— ) va servir à la synthèse de nouveaux brins (+) toujours grâce à la réplicase dans un complexe appelé “intermédiaire de réplication”. Ces nouveaux brins (+) vont être encapsidés afin de former de nouveaux virions. » Le virus s’assemble dans le réticulum endoplasmique, une structure en forme de réseau de canaux occupant l’espace dans le cytoplasme. Il est ensuite transporté par une vésicule vers la membrane plasmique qu’il traverse pour infecter une nouvelle cellule.

Sans l’ARN réplicase le virus n’a aucune possibilité de se reproduire dans le cytoplasme cellulaire. Elle est donc une cible pour des molécules thérapeutiques qui la bloqueraient ou en fausseraient le mécanisme de fonctionnement causant dans l’ARN viral des intercalations délétères de bases azotées chimiquement voisines, mais différentes des nucléotides lui faisant perdre sa capacité de coder des protéines virales fonctionnelles. La grande difficulté dans l’établissement d’une telle drogue est que la rendre spécifique à 100 % de l’ARN polymérase virale est extrêmement compliqué, voire pas envisageable. Il en résulte en pratique que toute molécule thérapeutique (comme le remdésivir) qui la ciblerait produirait également des effets délétères génotoxiques sur les cellules de l’hôte.
Partie 2 — Les intercalations erronées conduisent à des mutations du virus qui sont réparées par une enzyme spécifique, unique aux SARS-Cov1 et SARS-Cov2
La réplication des virus à ARN est très vulnérable à un nombre de facteurs, dont les enzymes de modification de l’ARN produites par le système immunitaire et les erreurs de copie dues à leur réplicase. L’intercalation d’une base azotée qui ne correspond pas au code génétique de départ a lieu. Ces erreurs arrivent fréquemment puisque le taux d’erreur intrinsèque de leurs réplicases est l’ordre de quelques erreurs par copie du virus, mille fois supérieur à celui des virus à ADN. Cependant, les virus ont développé une batterie de mécanismes pour compenser ce problème et augmenter considérablement la fidélité du processus de réplication de sorte que le taux d’erreur diminue radicalement. De plus, de nombreuses mutations sont silencieuses, car produisant le même acide aminé ou se produisent dans la partie non codante, ou conduisent à un virus non viable. Mais ces mutations restent immensément fréquentes au court d’une épidémie, puisque l’organisme d’une seule personne infectée peut produire des millions de virions (copies individuelles du virus), chaque cellule infectée de l’organisme en relâchant des centaines, voire des milliers, avant d’éclater ou de rentrer dans un processus apoptotique résultant de l’infection. Seules quelques mutations finissent par engendrer des mutations que l’on appelle « fixées » parce qu’elles dominent sur le territoire de propagation, car bien adaptées à l’hôte et à la contagiosité nécessaire à la survie du virus. Plus l’épidémie se répand et plus les chances d’obtenir des mutations très hautement pathogènes s’accroissent par le fait de la multiplication des individus atteints. Plusieurs mutations concomitantes peuvent être nécessaires pour augmenter certaines caractéristiques infectieuses. Il existe à présent plus d’une centaine de formes du virus par mutations fixées répertoriées ou comme variants* pour le SARS-Cov2 à travers le Monde. Certaines comme celles qui se sont répandues en Europe sont considérablement plus létales que les variants initiaux de Wuhan (certaines souches européennes ayant seraient 270 plus virulentes).

Dans le cas du SARS-Cov-2, l’ARN polymérase est dix fois plus rapide que celle des autres virus à ARN, mais beaucoup moins fidèle engendrant un taux de mutation beaucoup plus élevé. « Elle produit des erreurs qui, si elles n’étaient pas corrigées, mèneraient à une détérioration rapide du virus. En utilisant des médicaments antigrippaux comme l’Avigan, des chercheurs sont arrivés à lui faire accumuler de nombreuses mutations jusqu’à rendre le génome non fonctionnel. »
Cependant, cette faiblesse est compensée ce qui permet finalement à ce virus d’avoir une vitesse de réplication exceptionnelle sans que cela dégrade l’intégrité de son génome.
Isabelle Imbert, chercheur au CNRS, étudie le complexe de réplication du SARS Cov depuis 2003 à l’université Aix-Marseille. Elle est une spécialiste des coronavirus et l’équipe dans laquelle elle travaille au Laboratoire Architecture et Fonction des Macromolécules Biologiques (AFMB) de Marseille a découvert que la principale enzyme de réplication du SARS-CoV de 2003 (l’ARN polymérase) était assistée de deux cofacteurs indispensables (nsp7 et nsp8), c’est-à-dire des protéines participant au complexe de réplication.
« De plus, ces virus possèdent une enzyme exonucléase (la protéine non structurale nsp14) qui assure un “contrôle qualité” sur le génome, afin de corriger les erreurs qui peuvent se produire au cours de la réplication. “Cela explique la grande stabilité de la séquence d’ARN au cours du temps et entre les différentes souches d’une même espèce » nous dit-elle.
« Ce mécanisme apporte aussi des éléments de réponse quant à l’absence d’effet de la ribavirine chez les patients infectés par le SARS-CoV ou le MERS-CoV, alors qu’il s’agit d’un antiviral à large spectre, efficace par exemple contre le virus de l’hépatite C. “Ce médicament s’incorpore dans l’ARN du virus, à la place d’un nucléotide, et perturbe le fonctionnement du virus l’empêchant de se reproduire de façon viable. On parle d’analogue nucléotidique. Mais chez les coronavirus, il est expulsé par l’exonucléase”, explique-t-elle encore. » (Source : INSERM)
« Ainsi, pour la première fois, une explication est avancée quant à l’inefficacité de ce médicament contre les coronavirus. Tous les médicaments analogues de nucléosides qui fonctionnent sur ce principe en leurrant l’ARN polymérase sont susceptibles d’échouer également. » (Source : Theragora).
Il faut noter au passage que le remdésivir de Gilead dont le principe d’action est basé sur le fait que son métabolite actif GS-5734, est analogue de nucléotide, perturbe l’action de l’ARN polymérase et échappe à la correction des erreurs par l’exoribonucléase (Maria L. Agostini et al., « Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease », mBio, vol.9, no 2,mars-avril 2018). Du fait de la nécessité d’un dosage élevé et d’un manque certain de spécificité pour l’ARN polymérase virale (voir plus haut) il est également un médicament d’une toxicité avérée en particulier pour les reins.
Cette découverte contribue non seulement au développement de nouvelles stratégies thérapeutique, mais révèle que les coronavirus ont acquis eux-mêmes cette fonction de stabilité génomique. En effet l’exonucléase impliquée possède un repliement unique ce qui indique qu’elle n’a pas été acquise par recombinaison avec un autre virus, mais développée par les coronavirus au cours de leur évolution. Cette propriété structure/fonction montre que les coronavirus se situent à un des points de bascule de l’évolution pour la stabilité génomique.
Il semble donc que les voies de la Nature soient tout aussi impénétrables que celles de Dieu. La destruction ou la perturbation de l’habitat de l’espèce mammifère la plus reculée et la plus sauvage, héritière de l’ère des dinosaures, était une première menace comme nous l’avons décrit au chapitre 3. Mais encore plus sûrement le trafic culinaire à grande échelle ainsi que les études universitaires GOF sur les virus qu’elle véhicule, sont autant de facteurs qui ont conduit, ou vont conduire inexorablement, à la libération de virus au potentiel extraordinairement pathogène pour l’espèce humaine. Par sa technologie et son pouvoir de dévastation, l’Homme est devenu l’espèce dominante de la planète bien avant les insectes. Il a proliféré au-delà du raisonnable, pas tant en nombre d’individus, mais par son insatiabilité à détruire son environnement. La mise en danger de la planète par un désir de profit confinant à la folie (quel intérêt à mettre à son menu des chauves-souris) finit par déclencher un mécanisme écologique de régulation des espèces proliférantes.
Partie3. Les mutations et leur fréquence jouent un rôle crucial dans le déroulement d’une épidémie
Une très grande confusion règne dans les médias, car de nombreuses informations sur le SARS-Cov2 et l’évolution de l’épidémie s’entrecroisent de façon contradictoire. Par exemple certains intervenants font état d’un virus qui mute pendant que d’autres affirment que le virus ne mute pas, chacun étant persuadé de détenir la vérité. Cette cacophonie résulte du fait que chacun répète une chose différente entendue incidemment ou reçue après interview d’un expert scientifique. Malheureusement, les experts scientifiques ne sont en général pas de bons communicants parceque’ils n’essayent pas de rendre intelligible ce qu’ils disent et se fourvoient souvent sur la façon dont leurs dires vont être déformés en étant répétés.
En ce qui concerne les mutations : tous les virus mutent. Un virus qui ne mute pas n’est pas un virus. La seule question est de savoir à quelle vitesse il mute et qu’elles sont les parties de son génome qui mutent. Mais parler de vitesse de mutation prête également à confusion, malheureusement, pour la compréhension des journalistes et celle du grand public, car la vitesse de mutation n’est que le reflet du nombre total de cycles de réplication du virus sur l’ensemble d’une population infectée. Un virus contenu dans un tube à essai va y rester des décennies sans muter puisqu’il ne se réplique pas.
Un virus pandémique infecte des centaines de millions d’individus en se répliquant dans chacun d’entre eux de très nombreuses fois, des millions de fois en fait s’il n’est pas stoppé à temps par le système immunitaire. Comme nous l’avons vu les mutations sont causées par des défauts causés par la machinerie cellulaire de réplication tout comme une photocopieuse va engendrer quelques défauts presque invisibles dans un document. Si l’on procède mainte et mainte fois à la copie de la copie du document sa qualité va finir par diminuer et à la fin le texte photocopié de ne sera même plus reconnaissable.
Donc, en pratique les mutations ne sont que des changements parfaitement aléatoires à la base. Elles jouent pourtant un rôle critique bien connu dans les départs d’épidémie par l’émergence de virus mutants appelés souches pathogènes et contagieuses adaptées à l’hôte. Elles jouent également un rôle critique dans la fin des épidémies avec l’atténuation de la virulence résultant également de mutations délétères pour la survie du virus. De nombreux autres facteurs peuvent intervenir aussi de façon très complexe et très mal comprise dans la fin d’une épidémie : ils sont saisonniers, géographiques, alimentaires, sociétaux. Il faut noter que les virus peuvent également produire des variants* par recombinaison (rappariement) avec des virus semblables ou de même type en circulation endémique de faible pathogénie. C’est ce qui se passe d’ailleurs avec le SARS-Cov2 ou depuis l’été 2020 circulent de nouveaux variants plus ou moins pathogènes, chaque variant présentant d’ailleurs de multiples mutations.
Le virus initial dominant de Wuhan ne circule plus, mais d’autres variants et mutants en provenance de différents pays l’ont remplacé ce qui relance quelque peu l’épidémie. Dans chaque territoire le virus a évolué en mutant et s’est recombiné de façon indépendante avant d’être réexporté à l’occasion des échanges humains pendant la saison estivale. Tout cela indique à quel point, plus que les mesures au niveau individuel, le maintien de la fermeture des frontières est l’élément essentiel pour la prévention de la première venue d’une épidémie, mais aussi de sa réintroduction.
S’il est très facile de comprendre comment la pression évolutive entraîne le début d’une épidémie, il est plus malaisé de savoir comment elle peut contribuer à sa fin. En fait, il faut considérer que la pression sélective recouvre plusieurs aspects qui peuvent logiquement se schématiser comme suit :
(a) seules les mutations qui n’empêchent pas le virus de se répliquer et d’infecter un nouvel hôte perdurent ;
(b) les mutations qui augmentent la contagion prennent le dessus dans un premier temps puisqu’il y aura plus d’individus infectés transportant cette souche dans une population non immunisée ;
(c) les mutations plus pathogènes produisent plus de virus dans un même individu le rendant potentiellement plus contagieux, par le nombre de virus exhalés, mais le rendant très malade également plus vite. Un individu très malade réduit ses déplacements et le risque de contaminer un grand nombre de gens ;
(d) les mutations moins pathogènes permettent aux individus les plus résistants de ne pas développer de symptômes et du fait de leur activité sociale vont répandre le virus dans le reste de la population favorisant le développement de résistance immunitaire dans la population par l’intermédiaire de variant viraux moins pathogène, mais plus contagieux.
Les pressions évolutives (a) et (b) présentent un aspect intrinsèque et ont lieu quelle que soit la structure sociale de la population hôte. Elles sont à l’origine de l’adaptation d’un virus à une population et du départ d’une épidémie. En début d’épidémie, des mutations sont nécessaires pour que le virus s’adapte à son nouvel hôte lors du franchissement de la barrière interespèce, ce qui souvent nécessite l’adaptation intermédiaire à une espèce d’animaux domestiques ou de proximité avec l’homme.
Ensuite, pendant une phase transitoire qui peut durer plusieurs semaines ou plusieurs mois le virus continue de muter pour devenir de plus en plus contagieux et pathogène. La pathogénie est une résultante de l’adaptation de réplication. Plus son taux de réplication est élevé dans l’hôte et plus il est pathogène (virulent), car le système immunitaire est submergé et n’a pas le temps de réagir correctement, ou bien réagi brutalement et massivement entraînant le fameux orage de cytokines, une réaction immunitaire qui détruit les poumons et autres organes touchés par le virus, entraînant une mort quasi assurée alors que le virus a disparu du corps.
Les mutations virales correspondant aux pressions évolutives (b) et (c) sont au cœur des fantasmes et des poncifs des films hollywoodiens série B. Qui n’a pas vu la scène du chercheur en microbiologie, enfermé dans son scaphandre de protection niveau P4, qui observe au microscope un virus hautement pathogène et découvre, les yeux écarquillés, qu’il a muté ? Ce raccourci cinématographique a contribué à associer mutation à danger obligatoire dans la perception du grand public. Par contre la pression évolutive de fin d’épidémie ne fait l’objet d’aucun fantasme bien que ce soit une composante qui existe bel et bien.
La réalité de la pression sélective (c) peut varier considérablement en fonction de la structure sociale et de la contagiosité intrinsèque du virus de départ. Rappelons le rôle fondamental que joue depuis de très nombreuses années le transport aérien dans la propagation des grippes saisonnières. Le SARS de 2002 avait atteint le Hilton de Hong-kong d’où il s’était répandu vers Hanoï et une trentaine de pays à cause d’un médecin de Canton super-contaminateur (chapitre 3) qui avait passé une nuit très malade au Hilton.
L’époque moderne veut que l’on puisse tomber malade au cours d’un voyage qui nous amène en quelques heures à des milliers de kilomètres de chez nous. Certaines personnes parfois de façon totalement inconsciente, tentent de voyager même lorsqu’elles sont malades, à l’instar d’un cardiologue de l’Hôpital français de Hanoï (Arte, Pandémie : la traque planétaire — intégrale) qui pris de peur devant les symptômes de SARS qu’il était en train de développer avait décidé de rentrer en France pour s’y faire soigner sans le déclarer aux autorités françaises. Il a passé 10 heures avec 300 passagers dans un avion de ligne vers la France contaminant quelques passagers qui auraient pu à leur tour devenir des contaminateurs. Heureusement, aucun des passagers n’est mort et le SARS ne s’est pas répandu en France. Le médecin est décédé, à l’isolement, dans un service de réanimation qui fort heureusement avait été complètement séparé du reste de l’hôpital. De même, le Covid-19 s’est répandu comme une traînée de po enflammée autour des lignes de transport aérien
intercontinentales. De sorte que la pression (c) engendre à notre époque à coup sûr la propagation extrêmement rapide d’une épidémie, la transformant en pandémie.
La pression sélective (d) est également un mécanisme qui ne se met pas nécessairement en place, car il est tributaire de la vitesse de propagation de la souche la plus pathogène. Si les mutants les plus pathogènes circulent plus vite que les mutants les moins pathogènes alors l’épidémie ne s’arrêtera que lorsqu’elle aura rendu malade le plus grand nombre d’individus possible. Cependant, les courbes de décès et d’individus positifs, au moment de la mise en place du second confinement, semblent indiquer que nous sommes dans un processus de fin d’épidémie par pression évolutive de type (d). Étant donné le décalage entre la contamination et le décès possible il faut attendre une ou deux semaines pour en avoir la confirmation.

Les prédictions alarmistes que l’on a entendues au sujet de 500 000 morts au moment de la première vague et de 400 000 pour la deuxième vague ne tiennent aucun compte de l’évolution du virus en fonction des mécanismes de pression sélective. Elles se basent uniquement sur le taux de réplication R0 (entre 3,6 et 4) établi sur les données du début d’épidémie à Wuhan. De plus, le chiffre avancé pour la deuxième vague prend en compte que seulement 20 % de la population française a été en contact avec le virus au cours de la première vague. Ce dernier chiffre qui correspond à 13 millions de personnes se base sur le fait que 4 millions d’entre elles ont consulté pendant la période du confinement de mi-mars au 12 mai, pour des symptômes reliés au Covid-19 et qu’un grand nombre de gens disent avoir eu des symptômes légers avant le confinement sans compter tous ceux qui ont été asymptomatiques ou n’ont simplement pas été contaminés par le virus malgré des contacts avec une ou plusieurs personnes positives.
Rappelons que sur le porte-avions Charles de Gaulle où les marins ont été confinés pendant 1 mois complet entre fin février et début avril, dormant dans des chambrées ou l’atmosphère est en principe entièrement filtré et recyclé ce qui a peut être aidé à une large contamination. 30 % des personnels n’étaient pas positifs et 1/4 du reste de l’équipage qui était positif au Covid-19 était asymptomatique ou n’avait pas eu les symptômes classiques. « Sur les 1.568 marins, aviateurs et autres personnels du bord, 1.064 ont été diagnostiqués “positifs”, soit 69,7 %. Sur les 1.064 contaminés, les asymptomatiques représentent 13 %, 8 % des malades ont développé des symptômes atypiques : ni fièvre ni toux ni perte du goût ou de l’odorat, mais des douleurs thoraciques ou des maux de tête. » (Source : FranceInter)
Rappelons également que durant la peste noire de 1720, Marseille avait été isolée par un cordon sanitaire militaire et plus personne ne pouvait y rentrer ou en sortir. 50 % des habitants sont morts alors que la létalité de la peste est de 70 %. Donc, 30 % des habitants malgré le confinement de la ville n’ont tout simplement pas développé de symptômes. À cette époque il n’y avait d’ailleurs pas confinement individuel, sinon on n’aurait pas pu ramasser les morts.
Rappelons enfin que pour l’instant, cette épidémie, que nous jugeons dramatique, n’a causé officiellement en France que le décès de 0.05 % de la population (0.013 % dans le Monde) sur 10 mois alors que meurt environ 1 % de la population chaque année en France. La France a déploré pendant le confinement le décès de 31 000 personnes, essentiellement des personnes très âgées, ou à risque en raison du cumul de pathologie (diabète, obésité, hypertension, cancer évolutif). Le chiffre de 500 000 avancé sur 20 % correspond à 100 000 décès. Le confinement général de la population de la mi-mars au début mai aurait donc évité le décès de 69 000 personnes. Cela est n’est pas crédible par rapport au nombre de décès en Suède. La Suède où n’a été imposé aucun confinement se retrouve à la fin du mois d’octobre 2020 avec la même proportion de décès (5900 pour 10,3 millions d’habitants).
Il est vrai que la population la plus à risque est celle des personnes âgées en EHPAD et qu’elle doit bénéficier de mesures de protection qui peuvent être prises en dehors d’un confinement généralisé. Il n’est pas croyable d’entendre de la bouche du Président de la république qu’il serait injuste d’imposer un confinement à certains groupes de personnes et pas à d’autres et que de ce fait tout le monde doit se confiner en achevant d’éteindre l’économie déjà trop précaire de la France. Le principe de la République française est l’égalité des chances pas celle des malchances.
En s’équilibrant entre elles ces pressions évolutives sur les mutations conditionnent le déroulement d’une épidémie au fur et à mesure que la proportion de population contaminée augmente développant une immunité de groupe. En début d’épidémie dans les premières semaines ou mois on observe un renforcement du pouvoir contagieux (b) et de la pathogénie (c). En fin d’épidémie ce sont les mutations moins pathogènes qui prennent le dessus (d).
Les 4 types de pressions évolutives sont les paramètres d’un modèle simplifié de la réalité, mais ils permettent de se rendre compte de la complexité de la pression évolutive sur le développement d’un virus dans une population qui va au-delà de la modélisation mathématique basée sur le R0 et le R0 modifié par le confinement et le port du masque, prise en compte par le Haut Conseil de la santé publique.
Une étude théorique scientifique très sérieuse qui tente d’intégrer dans le modèle mathématique la pression évolutive a été publiée dans la revue Nature. Il s’agit de l’étude allemande de Sten Rüdiger de l’Université Humbolt de Berlin. La première phrase du résumé de l’article nous explique que : « Les épidémies et l’évolution de nombreux agents pathogènes ont lieu sur des échelles de temps similaires de sorte que leurs dynamiques sont souvent interreliées. » Bien entendu, cette étude publiée en 2020 s’applique tout particulièrement aux coronavirus qui ont un taux de mutation rapide. Dans une longue et passionnante discussion de conclusion de ses travaux, l’auteur explique, entre autres, que ses résultats indiquent que pour endiguer la diffusion d’une épidémie, il faut empêcher en premier lieu les mutations super-critiques (c.-à-d. extrêmement contagieuses) de se produire en coupant les communications à longue distance, c’est-à-dire les transports aériens. Nous voyons à quel point l’OMS et nos gouvernants ont refusé de suivre ce que par ailleurs la logique ancestrale préconise. En effet, nous explique-t-il, lorsque l’on donne de l’espace à un virus, non seulement le nombre de mutations explose avec le nombre de personnes infectées, mais le transport du virus vers une zone non déjà contaminée favorise l’émergence de mutants et variants plus pathogènes échappant à la compétition d’autres mutants et variants déjà présents. C’est ce qui s’est vraisemblablement passé entre Wuhan et Bergame (Italie) et ensuite entre Bergame et Mulhouse-Colmar (chapitre 4, partie 3). La ville de Bergame a vu l’apparition d’un virus dramatiquement plus létal que celui de Wuhan qui a probablement diffusé vers Mulhouse-Colmar et également vers les USA à la mi-février.
L’autre conclusion de l’étude de Sten Rüdiger est qu’une fois une mutation super-critique établie, comme cela s’est produit en Italie et en France (et probablement de la même façon dans bien d’autres pays comme l’Angleterre), il faut simplement isoler les zones géographiques de départ de ces mutations super-critiques de façon plus locale comme lorsque Marseille fut coupée de la Provence, et la Provence du reste de la France, pendant l’épidémie de peste à Marseille 1720. Dans cette étude aucune mention n’est faite du confinement individuel comme arme de lutte contre la propagation d’une épidémie…
Afin d’ajouter une note d’humour final à ce texte démoralisant, mentionnons l’article paru dans Futura le 31 octobre 2020 dont le but, à visée politique, est de nous enfoncer dans la tête, une bonne fois pour toutes, qu’il est civique de pratiquer la distanciation sociale par confinement individuel pour contrer la maladie. Cet article relate une étude « anthropomorphique » faite au Bélize par des chercheurs qui ont démontré que les chauves-souris pratiquent la distanciation sociale lorsqu’elles sont malades. Ils ont rendu malades 31 chauves-souris capturées en leur injectant une molécule destinée à affaiblir leur système immunitaire. Ils ont également muni chacune d’une petite balise GPS. Ils les ont ensuite relâchées dans leur colonie d’appartenance et ont pu mesurer qu’elles se tenaient à l’écart de leurs congénères dans les relations sociales, mais pas au cours de la chasse nocturne. Nous faisons remarquer à l’aimable journaliste scientifique qui rapporte cette recherche qu’il est observé depuis belle lurette que tous les animaux malades, ainsi que les humains, ont tendance à se tenir en retrait des interactions sociales lorsqu’ils se sentent malades. C’est un réflexe naturel qui permet tout simplement de se reposer en attendant de reprendre des forces. Par ailleurs, les chauves-souris malades arrêtent la distanciation lorsqu’il s’agit de se nourrir… quoi de plus naturel, tout comme les humains malades continuent d’aller au travail parfois malgré la maladie au détriment d’ailleurs de leurs collègues professionnels.
Le port du masque et le confinement sont donc venus interférer dans des mécanismes de propagation complexes pendant l’épidémie de SARS-Cov2 en France, sans que l’on sache réellement si cela a eu un effet bénéfique ou pas. Ces mesures ont imposé une pression évolutive dont on ne mesurera pleinement la réalité qu’à la toute fin de l’épidémie. Mais deux choses sont sûres : (a) on n’empêche pas un virus de proliférer à partir du moment où on lui donner accès à un territoire ; et (b) le confinement change la pression évolutive. En rallongeant l’épidémie, le confinement favorise à long terme les rappariements de virus et leur réintroduction ultérieure au moment où il se relâche par nécessité et que les frontières s’ouvrent à nouveau. Il n’est pas interdit d’imaginer que cette pression évolutive forcée puisse engendrer une alternance interminable de périodes de confinement et de déconfinement. Nous invitons les infectiologues désireux de donner leur avis sur cette situation complexe ou simplement désireux de réfuter cette hypothèse à publier une réponse dans France Soir.
Notes :
mutation : changement dans la séquence nucléotidique au niveau de l’ADN ou de l’une des paires de bases ; un virus mutant se caractérise par une ou plusieurs mutations critiques qui affectent notablement son pouvoir pathogène ou infectieux.
variant : changement dans la séquence nucléotidique comme des insertions, des délétions, tout réarrangement génétique. La plupart du temps ces changements conduisent à des propriété d’infectiosité et de pathogénie différente. Parfois le changement d’infectiosité est dû à un facteur environnemental.
Chapitre 6
Partie 1 — Un hôte intermédiaire est-il systématiquement nécessaire dans l’apparition d’une zoonose ?
Au chapitre 1, nous avons montré que la désinformation savamment entretenue par le gouvernement chinois, relayée par l’OMS et les gouvernements occidentaux, empêchait de connaître le point de départ exact de l’épidémie. La destruction des quelques 515 échantillons environnementaux et 70 échantillons d’animaux prélevés sur le marché humide de Wuhan empêche toute conclusion sûre sur le processus de franchissement de la barrière des espèces entre la chauve-souris et l’Homme. Les deux déclarations officielles contradictoires de Gao Fu, le directeur du Centre de Chinois de Contrôle des Maladies Infectieuses, à quatre mois d’intervalle, ne peuvent être considérées comme fiables. L’enquête circonstancielle directe n’est pas complètement forclose. Il reste toujours à cette heure la possibilité d’enquêter auprès des premiers malades de novembre 2019 qui ont survécu, ou auprès de leur famille, pour savoir si oui ou non ils ont été en contact avec un des marchés aux animaux sauvages de Wuhan, ou avec des animaux sauvages en dehors d’un marché, ou avec des personnes présentant des symptômes bénins de cette maladie. Devant le peu de coopération de la
Chine, il ne reste plus que la sagacité scientifique et l’analyse des génomes des coronavirus humains et de pangolins, saisis par les douanes chinoises, pour essayer d’y voir plus clair sur le mécanisme d’émergence du SARS-Cov2. On sait indiscutablement qu’entre 2017 et 2019 des pangolins de contrebande en provenance du Vietnam étaient été infectés de plusieurs virus, principalement par une lignée du virus Sendaï d’origine humaine ainsi qu’au moins deux coronavirus à SARS. Ces derniers se sont révélés par la suite être les deuxième et troisième virus les plus proches du SARS-Cov2. C’est la mine de cuivre abandonnée de Mojiang qui recelait en 2012-2013 le virus le plus proche du SARS-Cov2, connu à ce jour. Deux dates clés, 2012 et 2018, apparaissent autour d’un scénario possible de la genèse du virus qui reste un mystère. L’adaptation parfaite à l’homme du motif de liaison de la protéine S du virus du pangolin de 2019 est compatible avec un forçage adaptatif dans un primate, tels le chimpanzé ou l’orang-outan, qui aurait eu lieu courant 2018.
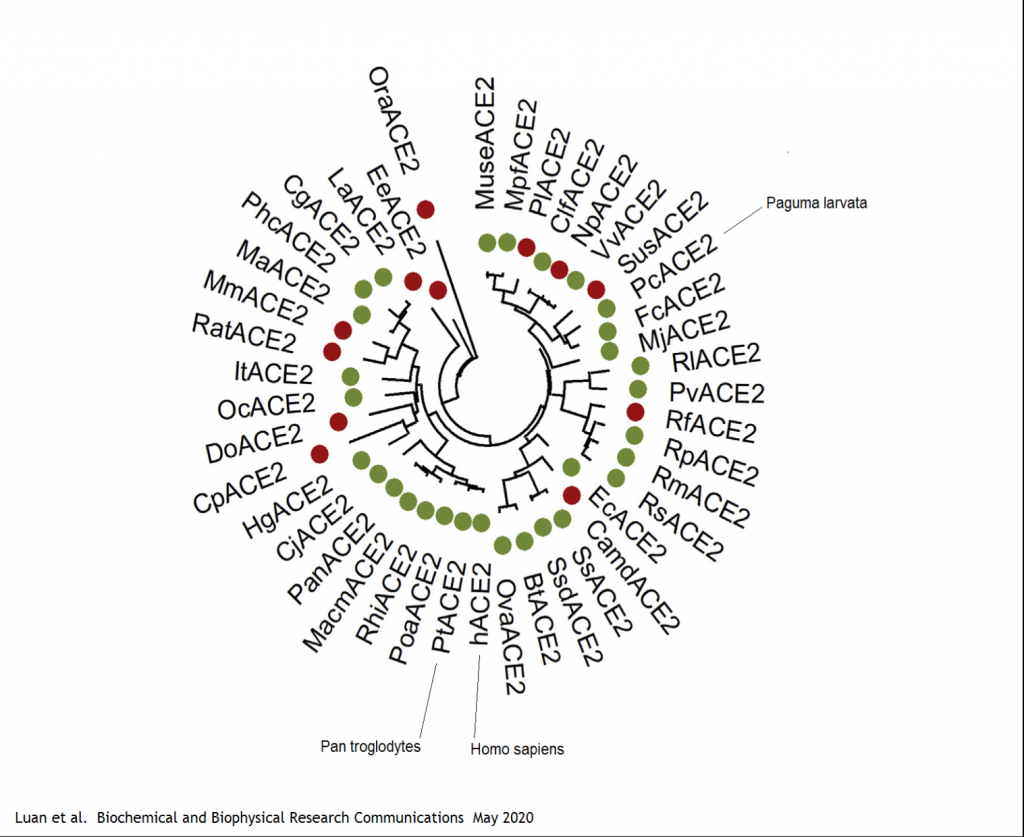
Les premiers coronavirus détectés chez les animaux familiers et l’Homme
Les coronavirus peuvent infecter des animaux domestiques et sauvages ainsi que les êtres humains. Les épidémies apparues au cours des 15 dernières années soulignent la capacité des coronavirus à franchir les barrières d’espèces pour causer des pathologies graves chez l’homme et montrent que cette famille de virus constitue un réservoir de pathogènes émergents. Selon les cas, ils provoquent des maladies respiratoires, digestives, hépatiques, neurologiques, de différentes sévérités.
Le premier coronavirus fut identifié aux États-Unis en 1931. Il s’agissait de celui de la bronchite infectieuse aviaire. Dans les années 1960, les premiers coronavirus humains furent isolés sur tous les continents, ils étaient peu pathogènes, responsables de rhumes le plus souvent anodins.
Chez les animaux domestiques, la plupart des coronavirus identifiés sont des alpha-coronavirus responsables de maladies graves comme le coronavirus du chat qui entraîne une péritonite infectieuse (PIF du chat) ou la gastro-entérite transmissible (GET) du porcelet. Le pronostic de la péritonite infectieuse féline est presque toujours fatal. Son origine est peu claire. Elle serait due à une mutation suspectée, mais non prouvée, du coronavirus entérique félin FECV/FeCoV bénin.
En 1984, l’émergence du coronavirus respiratoire porcin semblait avoir pour origine une modification du virus responsable de la GET (Cheng et al. 2007). La GET est devenue plus rare, parallèlement à l’émergence d’un autre coronavirus (Swine acute diarrhea syndrome coronavirus ou Sads-CoV), responsable de la diarrhée épidémique porcine (DEP), surtout importante dans le Sud-Est asiatique depuis 2010 (Sun RQ et al 2012). En 2017, une importante épidémie chez les porcelets permit de démontrer que le réservoir animal de la DEP était vraisemblablement la chauve-souris (Gong L et al 2017)
Les 7 coronavirus humains recensés jusqu’à présent
Avant 2003, seuls deux types de coronavirus, HCoV-229E et HCoV-OC43, étaient connus comme pouvant infecter les humains, provoquant principalement des infections respiratoires légères.
En octobre 2002, le premier coronavirus hautement pathogène émergea dans la population humaine. Il s’agissait d’un beta-coronavirus (le SARS-Cov), responsable de ce qu’on allait nommer le Syndrome Respiratoire Aigus Sévère (SARS) qui débuta en Chine avant de se propager rapidement dans 35 pays du monde.
Depuis, deux autres coronavirus du même type ont été identifiés, infectant l’homme à une large échelle. Contrairement à ce qui est affirmé dans les médias concernant le caractère bénin des coronavirus usuels, 3 des 5 nouveaux virus identifiés depuis 2002 sont létaux et ont causé des épidémies. Les 2 restants peuvent être également très pathogènes dans certains cas.
« Le HCoV-NL63 a été identifié aux Pays-Bas, fin 2004, chez un enfant de sept mois atteint de bronchiolite. L’infection par le virus a été confirmée dans le monde entier et est associée à de nombreux symptômes courants. Les maladies associées comprennent des infections légères à modérées des voies respiratoires supérieures, une infection aiguë sévère des voies respiratoires inférieures, un croup (laryngo-trachéo-bronchite) et de la bronchiolite. » (Source Wikipedia)
L’autre virus, le HCoV-HKU1 a été détecté pour la première fois en janvier 2005 chez un homme de 71 ans, hospitalisé en raison de symptômes de détresse respiratoire aiguë et d’une pneumonie bilatérale confirmée par radiographie. Il était récemment revenu à Hong Kong après avoir séjourné à Shenzhen, en Chine. Ce virus est distinct des autres coronavirus humains et semble s’apparenter au virus de l’hépatite murine.
En 2012, un beta-coronavirus extrêmement létal est apparu en Arabie Saoudite, nommé Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Ce sixième coronavirus qui infecte l’homme est responsable d’une épidémie restreinte à la péninsule arabique. À la fin novembre 2019, 2494 cas ont été confirmés dont 858 décès (soit un taux de létalité de 34,4 % selon OMS).
Finalement, le SARS-Cov2, officiellement apparu en décembre 2020, est le septième coronavirus recensé.

La civette palmiste, hôte intermédiaire identifié pour la première épidémie de SARS de 2002-2003
La plupart des premiers cas observés de SARS sont apparus en octobre 2002 à Guangzhou (Canton) dans la province du Guangdong. Les personnes qui manipulaient des aliments ou qui travaillaient dans les marchés aux animaux vivants présentaient des taux d’anticorps anti-SRAS-CoV plus élevés que les personnes exerçant d’autres professions (CDC Guandong 2003, Xu R-H, 2004). Des blaireaux-furets chinois, civettes palmistes masquées et un chien viverrin étaient infectés par un virus étroitement apparenté au SARS-Cov (marché de Shenzhen, 7 et 8 mai 2003) (Guan Y, Science 2003) démontrant que ces marchés étaient une route de transmission interespèces sans que le réservoir naturel du virus puisse être identifié. Leurs séquences génomiques présentaient jusqu’à 99,8 % d’identité avec celle du SRAS-CoV humain.
Sur cette base, plus d’un millier de civettes avaient été abattues en janvier 2004 à Guangdong. Cependant, aucune preuve concluante ne suggérait que les civettes étaient les hôtes réservoirs du SRAS-CoV ou que les civettes dans leur habitat naturel étaient infectées par le SRAS-CoV. La difficulté à trouver des civettes sauvages, les problèmes de réglementation rendaient les études de terrain sur les civettes sauvages compliquées, sinon impossibles.
Changchun Tu et al. avaient mené une étude sérologique sur la prévalence des anticorps au SRAS-CoV chez les civettes des marchés et celles des fermes d’élevage. Des tissus intestinaux et des échantillons de sérum avaient été prélevés sur 56 animaux : 38 civettes de quatre fermes d’élevage de différentes régions de la province du Guangdong (10 de Zhuhai, 10 de Shanwei, 9 de Shaoguan et 9 de Qingyuan) et 18 civettes du marché aux animaux vivants de Xinyuan à Guangzhou.

Des échantillons de tissus intestinaux et de sérum de civettes ont été sélectionnés dans 41 fermes distantes de plus de 100 km les unes des autres dans la province du Guangdong. L’étude avait également inclus 47 échantillons collectés en juin 2003 dans deux fermes dans les villes de Luoning (Henan) et Changsha (Hunan). Les conditions d’élevage dans ces fermes étaient similaires à celles du Guangdong. Sur 103 échantillons de sérum de civette testés, 18 étaient positifs à au moins un des trois tests utilisés, pour une séro-prévalence globale d’environ 17 % (aucune ferme n’avait une séro-prévalence > 40 %). Cette valeur différait considérablement de celles des marchés au gibier vivant de Guangzhou qui était de 78 % sur la même période. De plus, avec la distance la prévalence diminuait. Dans les 4 fermes de Shanwei, situées 240 km à l’est de Guangzhou, la séro-prévalence tombait à 10 %.
Ces résultats montraient que les civettes n’étaient pas le réservoir naturel du SARS-Cov, mais que l’épicentre de la maladie était bien le marché aux animaux de Guangzhou. Ce n’est que 10 ans après, en 2013, que Shi Zheng Li a pu identifier dans le Yunnan, à 1500 km de Guangzhou, deux virus de chauve-souris, Rs3367 et RsSHC014, partageant plus de 95 % d’identité avec les virus SARS-Cov humains et de civette (voir chapitre 4, partie 3). En tenant compte de l’évolution divergente des virus au fil des années, ces virus étaient mathématiquement directement apparentés au SARS-Cov. De même qu’il est mathématique que des chauves-souris vivantes, issues de trafics avec le Yunnan, devaient être présentes sur ce marché à certains moments avant le départ de l’épidémie. Cependant, les autorités chinoises restent très évasives sur la présence épisodique de ces animaux sur les marchés. De notoriété publique il s’agit là d’un secret de polichinelle. Un reportage de l’association PETA pour une éthique dans le traitement des animaux montre de façon indéniable des marchés d’animaux vivants au Cambodge, en Chine, en Indonésie, aux Philippines, en Thaïlande et au Vietnam, où des poulets, des canards, des poissons, des chiens ainsi que des chauves-souris, des singes et d’autres animaux exotiques sont vendus.
Il faut noter à quel point il est étrange qu’à Wuhan les 70 échantillons d’animaux saisis à la fermeture du marché de Huanan le 1er janvier aient été détruits, fermant à tout jamais la possibilité d’enquête épidémiologique conclusive similaire à celle menée en 2003 et 2004. La raison invoquée est que ces échantillons étaient en trop grand nombre pour être conservés en sécurité, vu la dangerosité du virus. Pour une ville possédant un laboratoire P4, cela s’appelle se moquer du monde.
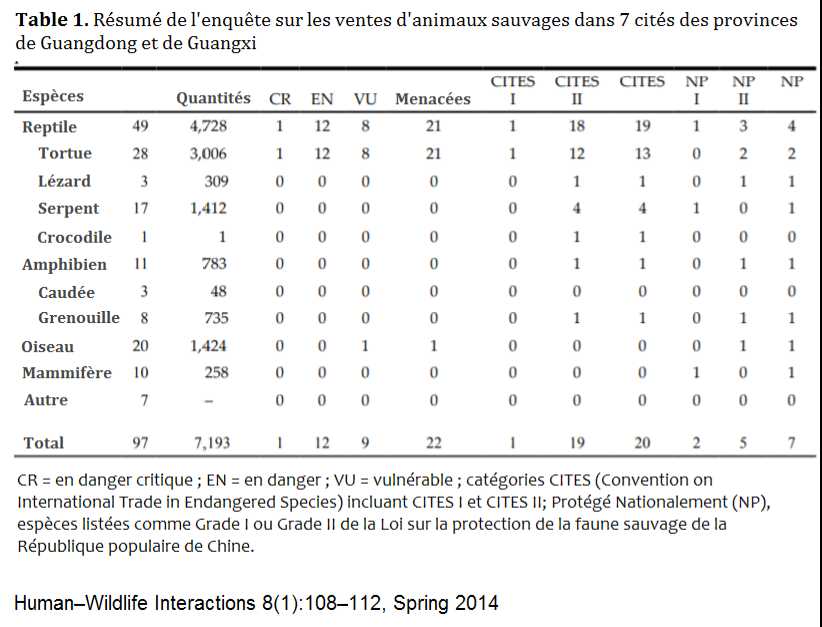
Le dromadaire, réservoir intermédiaire identifié pour l’épidémie de MERS de 2002-2003
En 2012, un nouveau virus respiratoire a fait son apparition en Arabie Saoudite. Baptisé MERS-CoV pour Coronavirus du Syndrome Respiratoire du Moyen-Orient, il touche le tractus respiratoire et est responsable de fièvre et de toux, pouvant entraîner la mort dans environ 30 % des cas. Le virus est alors détecté dans plusieurs pays du Moyen-Orient (Jordanie, Qatar, les Émirats Arabes Unis). Fort heureusement, la transmissibilité interhumaine est très faible (R0 < 1.) ce qui empêche la propagation d’une épidémie dont la létalité serait très probablement plus élevée que celle du SARS-Cov2 établie autour de 26 % des personnes hospitalisées. Quelques cas ont été détectés en Europe, dont 2 cas en France en 2013. Mais la faible transmissibilité interhumaine peut changer du jour au lendemain par une mutation du virus. Le 20 mai 2015, un cas (super-contaminateur) provenant du Moyen-Orient est identifié en Corée du Sud, ayant contaminé au 16 juin 2015 indirectement 154 personnes, dont 19 mortellement. À l’heure actuelle, aucun traitement
spécifique ou vaccin n’est disponible contre ce virus. L’enjeu actuel est de contenir l’épidémie, puis
de poursuivre les efforts de recherche pour mettre au point un vaccin et un traitement.
Des anticorps spécifiques à la protéine S du Mers-CoV ont été identifiés dans 100 % des échantillons de sérum prélevés sur une cinquantaine de dromadaires venant de différents endroits d’Oman, ce qui indique que le Mers-CoV, ou un virus extrêmement proche, circule largement au moins chez les dromadaires de la région. Des niveaux inférieurs d’anticorps spécifiques au MERS-CoV ont également été identifiés chez 14 % (15 sur 105) dromadaires des îles Canaries où la circulation du virus n’a pas été identifiée. Aucun anticorps spécifique au virus n’a été détecté chez les animaux étroitement liés au dromadaire.
Le MERS-CoV est capable de se répliquer dans différentes lignées de cellules de chauve-souris et son analyse phylogénique montre qu’il est proche d’un beta-coronavirus de pipistrelles (Pipistrellus) circulant en Europe et en Asie. L’horloge moléculaire des isolats humains du MERS-CoV évalue que leur divergence à partir d’un ancêtre commun s’est produite au milieu de l’année 2011, avec un foyer d’isolats dans la péninsule arabique divergeant à la fin 2012.
Quel hôte intermédiaire pour le SARS-Cov2 ?
En l’absence d’enquête et de données suffisantes fournies par la Chine, les recherches sur l’origine et l’évolution du SARS-CoV-2 viennent de l’analyse informatique des séquences génomiques associées à leurs métadonnées (date et le lieu de séquençage). L’Institut de France souligne que les conclusions que l’on en tire dépendent de modèles et d’approximations aussi bien que d’algorithmes souvent heuristiques (NDA, avec un caractère empirique qui permet de trouver, par exemple l’estimation de l’horloge moléculaire), du fait de la masse de données et la complexité des problèmes. L’Institut de France ajoute que : « Malgré ces limites, on a aujourd’hui des réponses très claires sur un certain nombre de questions, par exemple sur l’origine naturelle du virus et le fait qu’il n’est pas issu d’un laboratoire. » Nous nous proposons d’examiner minutieusement les données pour vérifier si cette affirmation s’étaye de preuves suffisamment concrètes pour exclure, a priori, un accident de laboratoire.
Certaines informations publiées sont contradictoires et nous devons faire attention aux déclarations officielles et universitaires véhiculées, qu’elles proviennent de la Chine ou de pays occidentaux. Le but de la Chine est évidemment de se dégager de toute responsabilité dans l’émergence de l’épidémie, de même que le gouvernement français cherche à occulter sa responsabilité dans la propagation de l’épidémie à la France. Et d’une certaine façon, il évite de demander officiellement une enquête sur les marchés aux animaux sauvages et un audit de l’Institut de virologie de Wuhan et son laboratoire P4. Nous avons traité au chapitre précédent de l’importance de la fermeture des frontières pour prévenir l’apparition et la réintroduction de variants hautement pathogènes.
Sur le plan des connaissances en virologie et épidémiologie, il n’est pas impossible que le SRAS-CoV-2 S ait pu développer naturellement un tropisme pour l’être humain directement chez les chauves-souris. Selon les travaux expérimentaux de Shi Zheng Li sur les virus synthétiques, publiés en 2015 (chapitre 2 et chapitre 3) cela est possible. On a vu en Australie, dans le milieu des années 1990, en deux occasions, le passage ultra rapide du virus Hendra de chevaux contaminés par des chauves-souris à l’homme, sur un période de temps très courte, sans réelle période apparente d’adaptation. Les chevaux sont morts et dans 2 cas sur 3 les hommes ont contact avec les chevaux aussi. Il n’y pas eu a priori d’adaptation évolutive du virus chez les chevaux. On a vu également des explorateurs imprudents mourir de la fièvre hémorragique causée par le virus de Marbourg, suite à la visite de la cave de Kitum au Kenya (dans laquelle un des médecins avait été contaminé également). Nous verrons dans la section suivante que des travailleurs envoyés pour déblayer du guano de chauve-souris dans la mine désaffectée de Mojiang dans le Yunnan, sont morts de toute évidence contaminés par un coronavirus de type SARS-Cov.
Cependant, à Wuhan on n’a pas de preuve de contamination directe par des chauves-souris ni de contamination indirecte ultra-rapide par le passage par un animal intermédiaire. Il serait donc nettement plus plausible qu’une adaptation graduelle se soit passée pour le SARS-Cov2 dans une espèce intermédiaire. En effet, l’étude théorique de la protéine de pointe S du SARS-CoV-2 montre qu’elle devrait se lier au récepteur ACE2 de pénétration cellulaire de très nombreuses espèces de mammifères. Elle se lie plus fortement que celle du SRAS-CoV à l’ACE2 de la chauve-souris et de l’homme. Selon l’étude théorique de Shang et al., parue dans Nature, la protéine S du RaTG13 (le virus de chauve-souris découvert en 2013 et proposé par Shi Zheng Li en février 2020 comme étant à l’origine du SARS-Cov de 2002) est également capable de se lier à l’ACE2 humain et, donc, a pu potentiellement infecter les êtres humains il y a 7 ans avant de se transformer.
Les calculs de l’horloge moléculaire du SARS-Cov2 indiquent que cela est possible (voir partie 3 de ce chapitre). Il est étonnant de constater que l’étude de Shang et al. a été citée par Alina Chan (chercheur-détective qui démontre brillamment que le SARS-Cov2 était déjà adapté à l’être humain en décembre 2019) pour affirmer que le RaTG13 n’infecte pas les êtres humains. Malheureusement, c’est une affirmation approximative qui contribue à détourner l’attention de l’Institut de virologie de Wuhan. En effet, on ne peut pas retrouver à présent le virus RaTG13 chez l’être humain puisque son identification remonte à 2013. Par contre, il s’agit d’un virus ancêtre possible du SARS-Cov2. Le RaTG13 ou un virus voisin a pu infecter des humains en 2012, comme nous le verrons dans la suite de notre analyse, mais il a forcément disparu aujourd’hui par la force de l’évolution. Le seul moyen de savoir qu’il n’a pas pu infecter les humains serait de le cultiver et le tester sur une lignée de cellules pulmonaires humaines, tout comme l’a fait Shi Zheng Li en 2015 pour prouver que son virus Covid chimérique synthétique avait les attributs d’un virus hautement pathogène (chapitre 3). Sur ce point nous sommes donc en désaccord avec Alina Chan.
Ce dont on est sûr scientifiquement, c’est que le séquençage a établi que le SARS-Cov2 a seulement 79,8 % d’identité nucléotidique avec le SARS-CoV (50 % avec le MERS-CoV), mais 96,2 % d’identité avec celui d’un virus de chauve-souris (Bat CoV RaTG13) et 90 % d’identité avec le génome d’un coronavirus détecté chez le pangolin. (source ANSES)
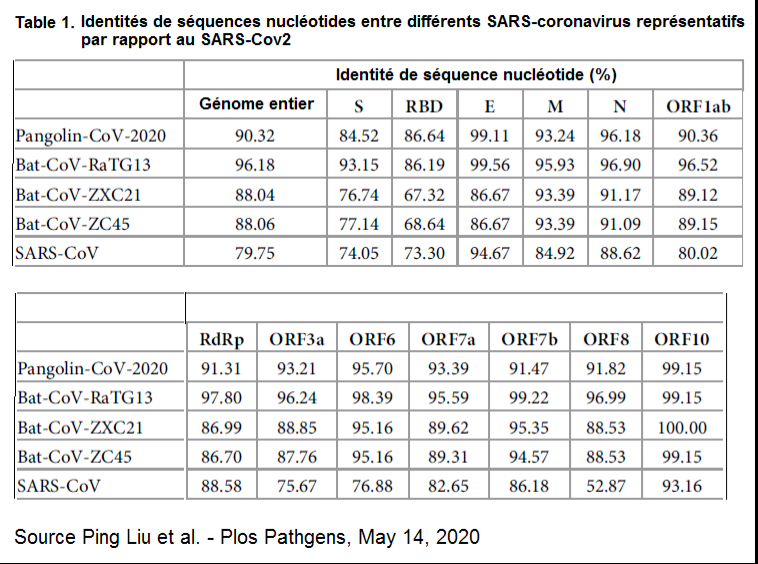
Partie 2 — Pourquoi le séquençage complet du virus RaTG13 n’a pas été communiqué par Shi Zheng Li avant février 2020 ?
Nous sommes obligés ici de faire une digression très importante à propos du virus RaTG13. Sa séquence déposée dans une banque de données internationale en février 2020 est arrivée à point nommé pour expliquer l’existence d’un ancêtre du SARS-Cov2 chez les chauves-souris. En effet, le SARS-Cov2 possède 96,2 % d’identité de séquence complète avec le RaTG13.
Cependant, quelques faits entourant le dépôt de la séquence du génome du RaTG13 posent problème. Tout d’abord, pourquoi avoir publié, en février 2020, le génome d’un virus provenant d’un échantillon prélevé en 2013 sur une chauve-souris (Rhinolophus affinis) dans la province du Yunnan ? Shi Zheng Li donne une explication en apparence simple et sans ambiguïté, dans un article publié dans Nature le 3 février 2020. Le génome complet du virus a été séquencé suite au fait qu’une séquence partielle de polymérase virale (RdRp), contenue dans l’échantillon RaTG13, (récolté en 2013 dans le Yunnan) matchait la séquence du virus extrait de patients en décembre 2019.
Cependant, il s’avère que les noms des fichiers de lecture des séquençages bruts du RaTG13, téléchargés vers la banque de données internationale GenBank, ont été trouvés par un membre anonyme de DRASTIC (Decentralized Radical Autonomous Search Team Investigating Covid-19) — une équipe d’enquêteurs indépendants sur le Covid-19. Les noms des fichiers indiquent que les expériences de séquençage se seraient étalées sur 15 mois, entre juin 2017 et septembre 2018. Nous pensons que les dates des fichiers sont fantaisistes par ce qu’en réalité le séquençage moderne d’un virus ne prend pas autant de temps. La Chine a annoncé la séquence du SARS-Cov2 dès le 13 janvier. Shi Zheng Li publie dans Nature qu’elle a séquencé le virus BaTG13 durant le mois de janvier suite au séquençage du SARS-Cov2. L’Institut Pasteur a séquencé le génome du SARS-Cov2 à partir de patients français infectés en 3 jours (en routine cela prend 10 jours). Donc, une opération de séquençage étalée sur 15 mois semble être une désinformation orchestrée pour entretenir le flou sur le date réelle du séquençage du RaTG13 à l’Institut de virologie de Wuhan. Cela permet peut-être de couvrir d’autres problèmes potentiels. Certains des quelque 28 collaborateurs de Shi Zheng Li, coauteurs de la publication dans Nature, ne pouvaient ignorer que les noms des fichiers officiellement déposés dans GenBank étaient en contradiction flagrante avec le texte de l’article accompagnant le dépôt de la séquence du RaTG13.
Répondant probablement à cette révélation, dans son entretien par e-mail avec le magazine Science, le Shi Zheng Li contredit ce qu’elle avait écrit dans son article paru dans Nature et affirme que le séquençage du génome complet du RaTG13 avait été effectué en 2018. Comment peut-on se « gourer » à ce point-là, dirons-nous avec de l’humour, car il en faut pour échapper au dogmatisme ambiant. Dans ce contexte de déclaration de collégienne, peut-on prendre pour argent comptant la déclaration que le RaTG13 n’a jamais été cultivé ?

Voici, traduite de l’anglais, la question posée par le correspondant de Science et la réponse de Shi Zheng Li :
« Que pouvez-vous nous dire à propos de la grotte [NDA, il s’agit en fait d’une référence à un incident dans une mine abandonnée] de Mojiang en 2012 ? Quand avez-vous isolé pour la première fois le RaTG13 ? Quand avez-vous achevé son séquençage complet ? »
Shi Zheng Li répond : « Nous avons détecté le virus par RT-PCR (avec des amorces couvrant largement les coronavirus) dans un prélèvement fécal de chauve-souris de 2013, provenant de la ville de Tongguan, dans le comté de Mojiang dans la province du Yunnan, et obtenu la séquence partielle de sa polymérase RdRp. En raison de la faible similarité de cette séquence avec celle du SARS-CoV, nous n’y avons pas prêté d’attention particulière. En 2018, comme la capacité de séquençage nouvelle génération (NGS) de notre laboratoire s’était améliorée, nous avons procédé au séquençage du virus en utilisant ce qui nous restait de l’échantillon. Nous avons ainsi obtenu la séquence complète du génome du RaTG13 sauf pour les 15 derniers nucléotides à l’extrémité 5’. Le virus RaTG13 n’a été détecté que dans un échantillon parmi tous les échantillons de chauve-souris collectés. En 2020, nous avons comparé les séquences du SARS-CoV-2 avec celle de notre coronavirus de chauve-souris non publié, RaTG13, et découvert qu’elles partageaient 96.2 % d’identité. Le RaTG13 n’a jamais été isolé ou cultivé. »
Pourquoi Shi Zheng Li n’a-t-elle pas publié la séquence du RaTG13 en 2018, elle qui a systématiquement accès au journal Nature, le journal scientifique le plus coté au monde ?
Étant donné le coût des nouveaux équipements de séquençage automatisé qu’elle mentionne dans sa réponse, la certitude de pouvoir publier dans un grand journal comme Nature et sa profession de foi, revendiquée, concernant la recherche et la prévention des zoonoses transmises par les chauves-souris, elle n’avait pratiquement aucune raison valable de retenir une telle séquence confidentiellement dans son laboratoire.
Soit jugeait-elle que cette séquence n’était pas d’une qualité suffisante pour être publiée, soit au contraire qu’elle était trop précieuse ou trop sensible pour être divulguée. Certains aficionados des théories complotistes pourraient se laisser aller à imaginer qu’elle pensait pouvoir l’utiliser à des fins de manipulations à gain de fonction (GOF). Une telle éventualité, sans intention de nuire, pourrait être crédible en raison du cursus avéré de Shi Zheng Li en matière de manipulations GOF (que nous avons présentées aux chapitres 2 et 3). Il faut comprendre que pour une savante de la classe de Shi Zheng Li il est important de garder la primauté d’une découverte potentielle. L’étude et la manipulation de la séquence d’un nouveau coronavirus peuvent permettre une nouvelle découverte et le risque de se voir doubler par un autre scientifique, dans le cas d’une publication trop rapide. Cependant, pour des raisons techniques il n’était pas possible que Shi Zheng Li ne puisse pas avoir le contrôle des découvertes reliées à cette séquence après son dépôt dans GenBank. La rétention de cette séquence complète entraîne donc des questions. Combien de génomes complets de coronavirus de chauves-souris, séquencés à l’Institut de virologie de Wuhan et son laboratoire P4, n’ont pas encore été révélés à la communauté scientifique internationale ? Il est indéniable que quand l’épidémie de SARS-Cov2 a éclaté à Wuhan, Shi Zheng Li s’est trouvée prise de court et a été obligée de publier immédiatement cette séquence dont d’autres personnes (chercheurs et techniciens) étaient forcément au courant. Un audit international scientifique de l’Institut de virologie et du laboratoire P4 de Wuhan s’impose. Mais la Chine n’est pas prête de l’accepter…
Mais il y a une autre raison très probable à la rétention par Shi Zheng Li de la séquence du RaTG13. Il est possible que le RaTG13 n’ait pas été publié par ce qu’il était en rapport avec un incident resté confidentiel de contamination zoonotique par un virus de type SARS-Cov. Cet incident, assez inquiétant en lui-même, qui a eu lieu en 2012 dans une mine à Mojiang, n’avait pas été déclaré à l’OMS pour des raisons qui restent obscures. Peut-être les tonnes de guano organique extraites avaient été vendues pour être épandues dans des champs en Chine ou exporter ailleurs pour être vendues très cher comme engrais de jardin à l’étranger ? Il faut se rendre compte que le trafic lucratif peut être à l’origine d’une épidémie. La peste de 1720 à Marseille est réputée avoir été introduite suite à la manœuvre des édiles de la ville pour éviter de perdre le bénéfice commercial d’une cargaison précieuse en provenance du Moyen-Orient, alors que des marins étaient morts de la peste à bord du bateau. Nous ne disons pas qu’une chose similaire s’est passée à Mojiang, mais cela montre qu’il faut rester extrêmement prudent vis-à-vis de toute ingérence dans le monde des chauves-souris, car les conséquences peuvent être incalculables. L’absence déclaration à l’OMS de cet incident est très fâcheux, car les autorités chinoises ont ainsi battu en brèche les efforts de surveillance internationale organisés pour prévenir les zoonoses du type SARS, dont la Chine est le foyer majeur.
L’incident de la mine de Mojiang
Dans son article d’investigation, publié le 12 octobre 2020 dans Changing Times, Annette Gartland donne le récit et les informations entourant l’incident de Moijang. Elles ont été déterrées par l’équipe DRASTIC qui montre qu’une base de données contenant des informations non publiées sur le séquençage d’échantillons collectés par l’Institut de virologie de Wuhan lors de voyages vers une mine de cuivre abandonnée du Yunnan a été retirée de l’internet.
On y apprend que six des hommes qui travaillaient dans la mine, enlevant les excréments de chauves-souris d’une grotte, ont souffert d’une grave maladie de type pneumonie en 2012. Trois d’entre eux sont décédés. Les mineurs avaient une forte fièvre, une toux sèche, des membres endoloris et, dans certains cas, des maux de tête — autant de symptômes qui sont maintenant associés au Covid-19.
Annette Gartland relate également des faits présentés dans un article publié par deux chercheurs scientifiques indiens, Rahalkar et Bahulikar. On découvre qu’un mémoire de maîtrise (en langue chinoise) a été trouvé sur le site Web cnki.net, décrivant en détail la maladie grave des mineurs. La thèse conclut qu’un coronavirus semblable au SARS-Cov provenant de chauves-souris (Rhinolophus) était l’agent causal prédit. Cette thèse, publiée en 2013, a été rédigée par le médecin chinois Li Xu, qui a soigné les mineurs et envoyé leurs échantillons de tissus à l’Institut de virologie de Wuhan.
Trois des mineurs sont morts dans un intervalle de 12 à 109 jours et trois ont survécu. La thèse comprenait des rapports médicaux, des images radiologiques telles que des tomodensitogrammes et des informations détaillées concernant le diagnostic et le traitement des mineurs. La radiographie a montré une pneumonie interstitielle et un syndrome de détresse respiratoire aiguë sévère (SDRA) chez certains patients. Certains ont montré des complications de la coagulation telles qu’une thrombo-embolie pulmonaire ou une thrombose.
La conclusion du Dr Zhong Nanshan (expert en maladies respiratoires et à présent conseiller national sur les épidémies de SRAS et de Covid-19) qui a assuré la consultation à distance des deux mineurs le plus gravement atteinte, était que leur pneumonie semblait être principalement virale et qu’elle était très probablement due à des coronavirus liés aux chauves-souris.
Les deux chercheurs ajoutent que, selon une traduction de la thèse de doctorat de Canping Huang : « les résultats des tests sanguins de quatre cas ont montré que : ces personnes portaient des anticorps IgG du virus du SRAS, dont deux qui étaient guéris avaient des niveaux d’anticorps plus élevés… et deux qui sont restés hospitalisés avaient des niveaux d’anticorps inférieurs… ».
Ils semblerait donc qu’un coronavirus à SARS proche du SAS-Cov2 ait pu infecter les mineurs. Les détracteurs de cette hypothèse avancent l’argument tout à fait faux que si les mineurs avaient été infectés par un coronavirus de cette nature, alors l’épidémie locale n’aurait pas pu être contenue. Rappelons que les mécanismes pré-épidémiques sont très mal connus et que les virus très pathogènes ne déclenchent pas forcément d’épidémie sur le coup. Les mineurs qui ont déblayé des tonnes de guano de chauve-souris dans la mine désaffectée de Mojiang près de la ville de Tuongguan ont baigné, au début d’avril, entre 4 jours et 2 semaines dans un aérosol de particules fines pénétrant leurs poumons. Ils ont été probablement contaminés par un virus très pathogène, mais extrêmement peu contagieux puisque personne d’autre n’a été touché dans le personnel médical. Ils ont commencé à faire face à des problèmes respiratoires, de la toux et de la fièvre qui ont nécessité une admission immédiate à l’hôpital de Kunming, fin avril et tôt en mai. Au chapitre 1, nous avons évoqué le virus de Marbourg, qui a causé la mort de quelques visiteurs imprudents de la grotte du Kitum et de 7 laborantins en Allemagne sans toutefois se transformer en épidémie. En ce qui concerne le MERS, quelques cas ont été détectés en Europe, dont 2 cas en France en 2013, sans créer d’épidémie. Cependant, l’absence de contamination peut être un phénomène très versatile, comme nous l’avons souligné précédemment.
Dans son article Annette Gartland mentionne qu’un membre de l’équipe DRASTIC, qui tweete sous le pseudonyme @ TheSeeker268, dit qu’en juillet 2012, quelques mois après l’épidémie de pneumonie chez les mineurs de Mojiang, il y a eu une opération de contrôle épidémique dans la zone de Tongguan qui a duré six mois.
Ce qui est certain c’est que le mystère et la désinformation ont une fois de plus régné sur cet incident. Rahalkar et Bahulikar s’interrogent : « Pourquoi la mine de Mojiang a-t-elle été visitée par des chercheurs jusqu’en octobre 2014 ? Des questions demeurent également quant à la raison pour laquelle le Dr Shi Zheng Li a attribué l’épidémie de Mojiang à une moisissure dans l’interview avec Scientific American. »
Nous avons traduit ce que dit exactement Shi Zheng Li dans son interview du 1er juin 2020 : « Le puits de mine puait terriblement » dit-elle, malgré le fait qu’elle portait une combinaison de protection et un masque. « Le guano de chauve-souris, recouvert de moisissure s’entassait dans la grotte » et elle ajoute de façon délectable : « Bien que la moisissure se soit révélée être l’agent pathogène qui a rendu malade les mineurs, ça n’aurait été qu’une question de temps avant qu’ils n’attrapent des coronavirus si la mine n’avait pas été fermée rapidement. »
« Tiens pardi ! » avons-nous envie de lui rétorquer. On ne peut pas plus se moquer du monde.
Nous ne sommes pas les seuls à douter, car Shi Zheng Li se défend en publiant, le 17 novembre 2020, un addendum à son article dans Nature où elle tente de démontrer que les mineurs n’ont pas été infectés par un coronavirus à SARS. Une fois de plus elle désinforme habilement. Mais elle est bien obligée de révéler que son groupe a collecté 1322 échantillons dans cette mine, entre 2012 et 2015. Ces échantillons ont permis d’identifier par PCR, sur la base d’une partie de la protéase RdRp, la présence de 284 alpha-coronavirus et 9 beta-coronavirus. Les 9 beta-coronavirus identifiés sont des coronavirus de type SARS-Cov. Rien que ça, avons-nous envie de dire. Donc elle admet, et même va plus loin en précisant qu’un de ces 9 virus est bien le RaTG13. Elle continue sa défense en précisant que la séquence partielle du gène RdRp du RaTG13 a été déposée à la GenBank en 2016, sous le code d’accession KP876546. C’est vrai, et le titre de l’article publié accompagnant ce dépôt est : « Coexistence de multiple coronavirus dans plusieurs colonies de chauves-souris dans un puits de mine abandonné. » Cela veut dire qu’en 2016 son laboratoire continuait de s’intéresser très activement au coronavirus à SARS que recelait cette mine.
Dans une autre partie de l’addendum, elle tente d’expliquer de façon très peu convaincante que les mineurs n’avaient pas été contaminés par un coronavirus à SARS. Elle explique de façon floue que 13 échantillons hospitaliers prélevés entre juin et septembre 2012, impliquant 4 mineurs dont un seul était mort, avaient été négatifs au virus Ebola et Nipah ainsi qu’au SARSr-CoV (en relation avec l’épidémie de 2003)… mais bien entendu cela ne voulait en aucun cas dire qu’ils n’étaient pas atteints d’un coronavirus encore inconnu. Comble de la désinformation, elle ajoute que les échantillons testés à nouveau cette année sont négatifs au SARS-Cov2 ! Le contraire aurait été surprenant étant donné que ce virus n’existait pas encore 2012, mais seulement un prédécesseur. On aurait donc préféré qu’elle teste les échantillons par rapport aux 9 SARS-coronavirus prélevés dans la mine.
Il n’est pas interdit de penser que les 8 autres SARS-coronavirus identifiés dans cette mine aient été séquencés entièrement à l’Institut de virologie de Wuhan, sous la responsabilité de Shi Zheng Li.
La dissimulation de la séquence du RaTG13 et les déclarations fumeuses de Shi Zheng Li à propos de la mine de Mojiang, ainsi que la non-déclaration de cet incident épidémique à l’OMS, sont autant d’aveux circonstanciels de rétention et de manipulation de l’information destinée à la communauté internationale. Si ces informations étaient diffusées dans les médias télévisuels, le grand public serait indigné et cela placerait la Chine dans une position diplomatique très inconfortable. Pour l’instant, elles restent dans un domaine confidentiel facilement assimilable à du « complotisme » et sont donc tout simplement ignorées. Ce mot générique permet de balayer d’un revers de manche des faits difficilement réfutables que nos élites ne souhaitent pas confronter en raison de leur conséquence politique.
Comme nous allons voir dans la partie suivante avec la théorie de l’horloge moléculaire, si l’on applique le rasoir d’Ockham*, en toute logique l’incident de la mine de Mojiang est la seule explication rationnelle à l’origine première de l’épidémie. Ce qui s’est passé ensuite ne pourra sans doute jamais être établi avec exactitude sans la coopération de la Chine. Que sont devenues les tonnes de guano extraites par les 6 mineurs ? Que sont devenus les échantillons prélevés par Shi Zheng Li sur des chauves-souris de Tongguan et ramenés à l’Institut de virologie de Wuhan ?
Partie 3 — La date clé de 2012 — quand et comment un virus apparenté au RaTG13 aurait-il pu passer à l’être humain et en combien de temps ?
Pour la plupart des virologues, le SARS-CoV-2 serait apparenté avec le RaTG13 par l’intermédiaire d’un ancêtre commun issu de la chauve-souris et le passage à l’humain serait récent. Il y a 3,8 % de différences entre les deux génomes, soit environ 1140 mutations, ce qui correspondrait entre 50 et100 ans d’évolution selon une fiche expert du 11 juin de l’Institut de France (Académie des Sciences) et une date comprise entre 1970 et 1995 pour l’ancêtre commun du SARS-CoV-2 et du RaTG13. Ce calcul se base sur un taux de mutation de 1 à 2 mutations par mois, mesuré en début d’épidémie (de début janvier à fin février), et un nombre de mutations approximé à 1200 (au lieu de 1138) par rapport au RaTG13. Ce calcul qui reprend celui de Bedford et Hutchinson publié en janvier manque de rigueur scientifique. La façon dont ce taux de mutations est établi n’est pas décrite. De plus, un taux calculé sur 2 mois, est réducteur de la réalité, voire très trompeur, car les recombinaisons (rappariements) entre virus qui sont une caractéristique essentielle des coronavirus ne sont pas prises en compte, ce qui fausse complètement l’estimation. Pour s’en rendre compte, il est primordial de comprendre comment fonctionne l’horloge moléculaire d’évolution des coronavirus de type SARS. Bien que d’une fiabilité relative, les horloges moléculaires apportent des indications précieuses, à la fois sur l’évolution de virus apparentés de façon très distance et sur la date d’apparition d’une lignée virale particulière dans une population. L’horloge spécifique de la lignée SARS-Cov2 permet d’établir de façon heuristique que sa date d’apparition se situerait entre fin octobre et mi-décembre 2019 (NDLR : typo corrigé le 2/12/2020). Ce résultat est cohérent par rapport aux cas identifiés rétrospectivement vers mi-novembre à Wuhan. Cependant, en Italie, une étude a détecté l’apparition d’anticorps spécifiques au SARS-Cov2 durant la période pré-pandémique, dès septembre 2019 dans la région de Lombardie, soit deux mois plus tôt. Étant donné que dans beaucoup de cas l’infection est asymptomatique ou pauci symptomatique, et que seulement 2 à 3 % des personnes présentant des symptômes sont hospitalisées (France), le virus a donc pu circuler de façon indétectable assez longtemps dans la période pré-pandémique.
Description de l’horloge moléculaire — le cas d’école du virus de la poliomyélite
Il faut savoir que l’établissement de l’horloge moléculaire d’évolution des espèces (incluant les virus) est une branche mathématique fondamentale de la science du vivant. Ses fondements remontent aux années 1960 avec les travaux d’Émile Zuckeerkandl, du prix Nobel de chimie Linus Pauling et du généticien japonais Motoo Kimura. Émile Zuckeerkandl et Linus Pauling remarquent en 1962 que le nombre d’acides aminés, qui diffèrent entre les hémoglobines des différentes espèces, change de façon approximativement linéaire avec le temps lorsque l’on classe les espèces par leur ancienneté d’apparition sur terre. Ils ont généralisé leur observation en formulant l’hypothèse de « l’horloge moléculaire », c’est-à-dire que le taux de mutations reliées à l’évolution de n’importe quel gène étaient approximativement constantes à travers le temps et les différentes lignées ou espèces.
Aujourd’hui, les méthodes automatisées de séquençage à haut débit permettent d’obtenir un très grand nombre de séquences d’un même virus qui se réplique et mute rapidement. On peut établir ainsi la phylogénie de l’épidémie et calibrer son horloge moléculaire avec une assez grande précision, mais pas forcément avec grande exactitude.
Il faut se rendre compte qu’il existe en réalité plusieurs horloges moléculaires qui décrivent les différents processus d’évolution d’un virus en fonction du type de mutation impliqué. Lorsque le résultat de la mutation ne change en rien la séquence en acides aminés, une fois les gènes transcrits en protéines fonctionnelles, la mutation est dite silencieuse ou synonyme. Lorsque la mutation induit un changement dans la composition en acides aminés d’un gène, la mutation est dite non synonyme. Parmi les mutations non synonymes, il y en a de deux types : celles qui vont influencer le fonctionnement du virus (pathogénie, contagiosité, durée d’incubation…) et celles qui n’auront pas d’effet remarquable. Les mutations qui changent de façon marquée le fonctionnement d’un virus, et lui permettent de se répandre dans une partie de la population, engendrent de ce fait une lignée nouvelle, appelée aussi variant.
Une étude très poussée sur les horloges moléculaires du virus de la poliomyélite, un virus à ARN qui comme le SARS-Cov2 se réplique très rapidement, a pu se faire et sert de référence en la matière. En 1981, un poliovirus importé du Moyen-Orient a supplanté un virus endémique de type 1 présent sur un territoire correspondant au nord des Andes (Venezuela, Pérou). Le virus importé a circulé largement jusqu’en 1991, année de son éradication. Le programme de surveillance a fourni des isolats viraux [NDA, un isolat est un représentant du virus contenu dans un échantillon] qui étaient en relation proche du virus importé et des isolats représentant deux principales lignées divergentes. L’analyse de toutes les séquences, par rapport à celle du virus initial, a permis d’estimer le taux de fixation (c.-à-d. taux d’apparition) des différentes catégories de mutations.
Ainsi, le taux Ka de fixation des mutations silencieuses était très rapide [(1.00 ± 0.08) × 10−2 mutations/site/an] et celui des mutations non silencieuses 30 fois plus lentes Ka [(0.03 ± 0.01) × 10−2] (c.-à-d., taux de mutation du gène long de 2.643 nucléotide qui encode les 4 protéines de la capside du virus qui ont une très grande importance fonctionnelle, car elles assurent la reconnaissance du récepteur de pénétration cellulaire CD155). Le taux global de mutations fixées calculé Kt [(1.03 ± 0.10) × 10−2] est par définition très proche du taux des mutations silencieuses qui sont très largement plus fréquentes que les mutations non silencieuses puisqu’elles impactent le virus d’aucune façon. Les horloges Ks et Kt permettent d’étudier la divergence de deux isolats épidémiologiques très voisins alors que l’horloge Ka estime le temps d’évolution entre des génotypes différents ou des variants possédant des propriétés infectieuses distinctes. Ces changements découlent d’une ou plusieurs mutations non silencieuses affectant des parties du génome avec une forte implication fonctionnelle, comme les protéines capsides du poliovirus ou dans le cas du SARS-Cov2 le domaine de liaison au récepteur ACE2 de la protéine S. En 10 ans, le
virus de la polio (d’une longueur d’environ 8 kb) introduit dans les Andes a donc accumulé plus de 800 mutations, soit 10 % de son génome, dont seulement environ 27 (0.3 %) se situaient sur la capside. La capside est doublement importante en raison de sa structure géométrique très complexe et de sa fonction de pénétration, d’où un Ka très faible traduisant une horloge d’évolution très lente, car la plupart des mutations dans cette région du génome sont délétères pour le virus.
De façon similaire, le ratio Ka/Ks = 0.054 (soit Ka 20 fois plus lent que Ks) pour la protéine S des coronavirus à SARS (SARSr-Rh-BatCoV) a été établi par Lau et al. en 2010, indiquant le caractère primordial de cette protéine pour la survie du virus chez les chauves-souris rhinolophes. Chez les chauves-souris, cette protéine est optimisée par l’évolution et de ce fait elle ne mute quasiment plus. Toute mutation de cette protéine entraînerait un affaiblissement de la capacité de pénétration cellulaire incompatible avec la survie du virus en compétition avec les autres souches virales au sein de l’espèce réservoir. Par contre, l’épidémie de SARS-Cov de 2002 a démontré que le ratio Ka/Ks était voisin de 1. pour les SARSr-CoV de civettes (1.5) et les SARSr-CoV humains (1.0), car le virus tend à optimiser sa pénétration cellulaire dans ces nouveaux hôtes.
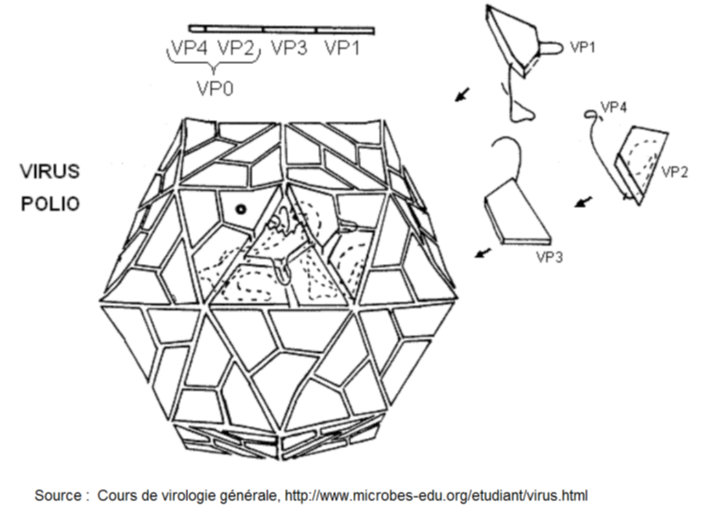
Un problème général en ce qui concerne les virus à ARN, comme le SARS-Cov2, qui évoluent rapidement est que les tendances évolutives sur une large échelle sont difficiles à cerner. C’est-à-dire, les relations de parenté entre des virus similaires, mais de génotypes différents, qui ont coévolué sur un large territoire et une très longue période de temps, sont difficiles à établir avec exactitude à cause de la méconnaissance de nombreux chaînons manquants. De plus, les virus se recombinent entre eux et avec d’autres d’autres virus de type différent. Il est donc impossible de reconstruire un arbre phylogénique complet les reliant ce qui entraîne l’inexactitude de la calibration de l’horloge moléculaire Ka fonctionnelle.
Estimation correcte de la date d’apparition de l’hypothétique ancêtre commun aux SARS-Cov2 et RaTG13
Premièrement, nous reprenons la méthodologie de l’Institut de France de façon plus rigoureuse en utilisant le taux global de mutations fixées Kt = 1.1 ± 0.4 × 10−3 mutations/site/an, calculé avec précision par Duchene et al. entre début janvier et le 24 février. Ce taux est 2 fois plus rapide que celui estimé par l’Institut de France qui l’avait estimé pourtant sur une même période. Soulignons qu’il est établi à un moment de l’épidémie où peu ou pas de variants de recombinaisons fixées ont émergé et que, compte tenu de la barre d’erreur, il indique que l’apparition du virus souche de l’épidémie humaine a eu lieu entre fin octobre et mi-décembre 2019.
Pour 1138 mutations (entre RaTG13 et SARS-Cov2) et une séquence de circa 30.000 nucléotides, nous obtenons un temps d’évolution situé entre 25 et 54 ans qui devrait être divisé par deux si l’on considère que les deux virus ont divergé à la même vitesse à partir du même ancêtre commun. Mais cela induit une erreur par ce que le virus RaTG13 a arrêté d’évoluer en 2013 (date de l’échantillon prélevé), ce que l’Institut de France a négligé en raison de leur estimation du temps d’évolution (50 à 100 ans) très supérieur à la différence temporelle entre l’apparition du SARS-Cov2 et le prélèvement. Mais avec des temps plus courts, nous devons ajuster plus finement le calcul (y + x = 25 ou y + x = 54 et y — x = 7, avec x et y étant les temps d’évolution du RaTG13 et du SARS-Cov2). Nous obtenons donc une date comprise entre 1990 et 2004 (au lieu de 1970 et 1995) pourl’apparition de l’hypothétique ancêtre commun.
Cependant l’horloge d’évolution utilisée dans ce calcul est établie sur l’évolution rapide d’une lignée virale sur deux mois et ne prend pas en compte la fréquence des rappariements ou réassortiments au long cours (sur des années) de variants proches, très importante chez les coronavirus de chauves-souris. Ce processus conduit nécessairement à une accélération de l’horloge moléculaire. Une idée du taux possible de fixation des mutations, prenant en compte l’évolution au long cours est donnée par l’article de Benvenuto et al., avec une valeur calculée de 6.58 × 10−3 mutations/site/an avec un intervalle de confiance de 95 % entre 5.2 et 8.1 × 10−3 mutations/site/an. Cette valeur est 2 fois plus élevée par rapport à la valeur très sérieuse de 2.82 × 10−3 obtenue pour l’ORF1ab (voir chapitre 5) au cours de l’étude écoépidémiologique qui fait référence en la matière, conduite par Lau et al. entre Hong-kong et la Chine, sur les migrations de chauves-souris rhinolophes (fer à cheval) et l’hétérogénéité de leur virus de type SARS.
L’estimation de Benvenuto et al. est probablement relativement approximative étant donné le faible nombre (n=3) de coronavirus connus proches de la lignée du SARS-Cov2. Il faut noter, en a parte, qu’ils restent étrangement extrêmement discret dans leur article au sujet des 3 virus les plus proches du SARS-Cov2, utilisés pour calibrer l’horloge évolutive. Ils ne se réfèrent pas à eux directement dans le texte et il faut consulter la figure décrivant l’arbre phylogénique qu’ils établissent pour apercevoir en petits caractères les noms de code GenBank de ces virus. Il s’agit du MG772933 et MG772934 et qui ne sont ni plus ni moins que des virus ZC45 et ZXC21, issus de la recherche militaire chinoise amplement décrite au chapitre 4 partie 3, le troisième virus étant le RaTG13.
Cette discrétion est évidemment conduite par la nécessité d’éviter que leur article soit repris et déformé par les sites d’information complotistes. Cependant, ce procédé maladroit des auteurs ne peut évidemment qu’attiser les sites complotistes, surtout que la dissidente de Hong Kong, Li Meng Yan, prétend tout simplement que le SARS-Cov2 a été fabriqué à partir des virus ZC45 et ZXC21. Cependant, nous allons voir plus loin cette hypothèse, bien que troublante, n’est pas étayée de preuves.
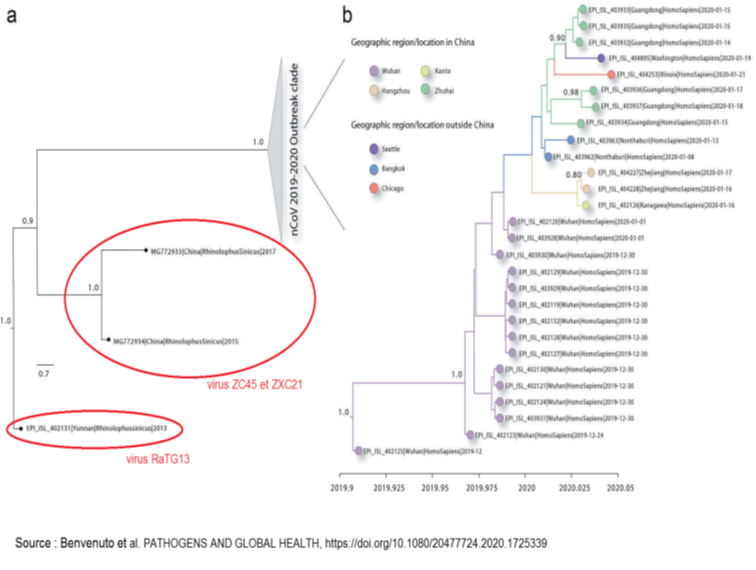
Il peut paraître étrange que des virus isolés par les chercheurs militaires à partir d’échantillon de colonies côtières de chauves-souris dans les provinces de Guangdong et de Fujian se retrouvent dans le calcul d’une horloge évolutive. On peut se demander évidemment comment il se fait que des virus identifiés dans des sites à 2000 km de la mine de Mojiang dans le Yunnan soient finalement relativement proches du SARS-Cov2. Mais n’oublions pas que le virus du pangolin est plus proche du SARS-Cov2 que les virus ZC45 et ZXC21. Il faut surtout comprendre que les seuls virus de type SARS à disposition sont le SARS-Cov, les ZC45 et ZXC21, celui du pangolin et le SARS-Cov2. Les militaires ont forcément séquencé en premier les virus de type SARS parmi les dizaines de coronavirus de chauves-souris qu’ils ont identifiés, car ce sont les plus dangereux.
Benvenuto ne donne que peu d’indications de la fiabilité de son résultat. Il se contente de dire que ses calculs sont auto-cohérents car le modèle mathématique particulier utilisé pour établir d’horloge est celui le plus cohérent avec l’ensemble de l’arbre phylogénique (coefficient de corrélation R2 = 0.85). Le fait que les virus militaires soient géographiquement très éloignés est un plus pour la fiabilité de l’horloge. Cependant, comme il le note, la proximité temporelle de l’origine des séquences militaires pourrait affecter le résultat et engendrer un biais d’accélération de l’horloge.
Les valeurs de l’horloge calculées par Benvenuto et al. indiquent que le temps d’évolution entre le SARS-Cov2 et le RaTG13 se situerait entre 7,3 et 4,7 années. Cela donne une fourchette entre 2012 et 2015 recouvrant exactement par sa borne supérieure l’incident de la mine de Mojiang. De ce fait, le RaTG13 ou un virus extrêmement proche pourrait bien être l’ancêtre du SARS-Cov2. La borne inférieure de l’intervalle est sans signification, car elle situerait l’ancêtre commun après l’apparition du RaTG13.
En utilisant la valeur obtenue par Lau et al., dont nous ne connaissons pas la barre d’erreur (non publié), nous obtenons la date de fin 2009 pour l’ancêtre commun du SARS-Cov2 et du RatG13. En tenant compte d’une marge d’erreur possible de 15 % sur la valeur de Lau et al., on obtient un intervalle de temps compris entre 2008 et 2011. Ce qui montre que dans l’hypothèse probable où la vitesse de l’horloge calculée par Benvenuto et al. serait surestimée nous obtiendrions un résultat qui continue d’indiquer le RatG13, ou un virus proche apparu entre fin 2008 et 2015, comme ancêtre commun du SARS-Cov2. L’incident de la mine de Mojiang en 2012 se trouve au milieu de cette fenêtre temporelle. Notons que l’étude de Lau et al. a montré que l’ancêtre commun au SARS-Cov de fin 2002 et du coronavirus de chauve-souris le plus proche remonterait à 1995. C’est-à-dire environ le même intervalle de temps (7 ans) qui sépare l’apparition du SARS-Cov2 de l’incident de Mojiang.
En conclusion, de toute évidence le séquençage en 2018 du virus RaTG13, dont pourrait certainement descendre le SARS-Cov2, n’a pas été révélé avant février 2020. De même, l’incident épidémique de la mine de Mojiang a été caché à l’OMS. Les dates des fichiers de séquençage et les déclarations de Shi Zheng Li dans son interview à Science contredisent ce qu’elle a publié dans le journal Nature. L’attitude et les déclarations de Shi Zheng montrent qu’elle s’est trouvée en février 2020 dans l’obligation de révéler en urgence au Monde l’existence du virus RaTG13. D’un autre côté, elle ne peut pas avouer directement qu’elle savait que les mineurs de Mojiang avec été contaminés par un coronavirus de type SARS, ce que tout dans leur tableau clinique indique. D’ailleurs, s’il n’avait pas été question d’une infection par coronavirus de type SARS pourquoi serait-elle allée à la mine collecter des échantillons fécaux de chauves-souris ? Pourquoi cet incident n’a pas été déclaré à l’OMS ? Quel rôle a joué Shi Sheng Li dans cette mascarade ? Où sont passées les tonnes de guano extraites de la mine ? Tout cela appelle une enquête internationale et un audit méticuleux de l’Institut de virologie de Wuhan et de son laboratoire P4.
Le virus a incubé dans la population pendant une longue période. Des preuves scientifiques indiscutables sont là qui montrent que le virus SARS-Cov2 était présent en Italie du Nord (Lombardie) dès la fin de l’été 2019. Durant l’année 2019, et plus particulièrement l’été, un nombre extraordinaire de touristes chinois (pas moins de 3,5 millions) ont débarqué en Italie du Nord. Les causes probables de cet engouement soudain pour l’Italie ont probablement été la guerre commerciale sino-américaine ainsi que les violences au cours des manifestations trop durement réprimées des gilets jaunes et les grèves à répétition en France. Cela veut dire qu’un ou des virus pré-pandémiques circulaient largement en Chine durant l’année 2019.
L’horloge moléculaire des coronavirus à SARS de chauves-souris indique qu’il ne faut que quelques années pour qu’un virus franchisse la barrière des espèces. La fréquence très élevée des réassortiments de coronavirus chez les chauves-souris en fait un réservoir sans cesse renouvelé de virus qui se comptent probablement par centaines ou milliers vu le nombre de colonies de chauves-souris en Chine. Les 10 000 marchés aux animaux sauvages et 20 000 fermes d’élevage de gibier, apparues ces 25 dernières années ont engendré une porosité sans précédent de la barrière des espèces. La Chine est devenue un incubateur géant à coronavirus à SARS par un mécanisme résonant d’amplification de contagiosité dû à des allers-retours de virus entre des animaux intermédiaires et l’être humain. Un exemple de ce mécanisme a été mis en évidence dans des fermes d’élevage de visons aux Pays-Bas, où le SARS-Cov2 est repassé de visons contaminés à l’être humain. Cette promiscuité omniprésente entre les chauves-souris, le gibier d’élevage et l’être humain qui n’est toujours pas contrôlée va augmenter la fréquence de transmission de zoonoses à l’homme si rien n’est fait dès à présent.
Partie 4 — Le pangolin — témoin et victime de la genèse de l’épidémie en Chine — n’est pas l’hôte intermédiaire
En avant-propos nous devons attirer l’attention sur l’annonce faite le 7 février 2020 que le pangolin était « l’hôte intermédiaire ». Elle a eu lieu deux semaines à peine après la révélation officielle de l’épidémie par l’OMS. Cet empressement, totalement infondé, n’est pas compatible avec le temps scientifique. Il s’agissait d’un temps de communication médiatique avec tout ce que cela implique comme erreur, cafouillage, absence de vérification, et surtout absence de réflexion et de recul, ce qui a finalement desservi le droit du public à une information vraie. Il avait fallu des mois d’enquête rigoureuse pour établir que la civette palmiste avait été l’hôte intermédiaire dans l’épidémie de SARS de 2002-2003. Seconde différence de taille avec les civettes palmistes élevées dans des centaines de fermes en Chine, le pangolin est un animal sauvage, rare en voie d’extinction. Il est introduit en Chine depuis le Vietnam par des contrebandiers en connexion avec des receleurs et des vendeurs sur les marchés aux animaux sauvages. Il est donc de ce fait totalement illogique d’en faire a priori l’animal-hôte intermédiaire. On ne sait même pas si parmi les 70 échantillons d’animaux prélevés sur le marché du Huanan à Wuhan se trouvait le pangolin. Comme nous allons le voir, le pangolin est en fait un témoin précieux de ce qui a pu se passer.
Dans leur annonce du 7 février, les deux chercheurs de la South China Agricultural University à Guangzhou ont affirmé que la séquence du génome d’un beta-coronavirus extrait d’un pangolin était 99 % « similaire » à celle du SARS-Cov2. En parlant d’un degré de similarité au lieu d’identité ils biaisent la réalité de l’annonce, car cette imprécision laisse planer un doute sur la signification de similaire. Le degré exact d’identité de séquence de 90,1 % au niveau du génome complet fait du virus du pangolin le deuxième virus le plus proche du SARS-Cov2 derrière le RatG13 (96,2 %). Cependant, au niveau des protéines transcrites par les gènes, l’identité globale monte à 96 % et même entre 98 et 100 % pour les protéines structurales, à l’exception de la protéine de pénétration S (90,2 %). Ces caractéristiques le rapprochent énormément du SARS-Cov2. Comment se fait-il alors qu’à l’exception de la protéine S, il soit moins proche du SARS-Cov2 que le RaTG13 qui est plus éloigné dans le temps ? Il s’agit là d’un vrai paradoxe évolutif.
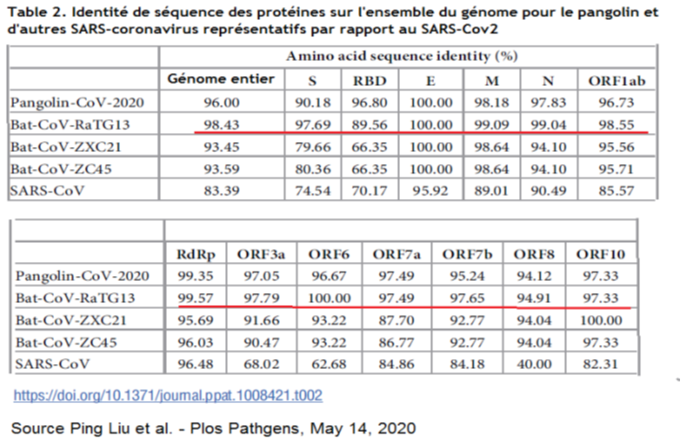
D’animal de contrebande au statut d’hôte intermédiaire dans la genèse de l’épidémie
Malheureusement pour eux et pour notre planète, les pangolins sont une espèce animale victime du snobisme culinaire chinois comme quelques autres espèces en voie d’extinction. L’odyssée médiatique de ces pauvres pangolins a commencé le 24 mars 2019, lorsque 21 pangolins javanais (Manis javanica) ont été saisis dans une opération des douanes chinoises, puis transférés malades vers le centre de sauvetage des animaux sauvages de Guangdong. Les premiers indices rassemblés par les agents des douanes suggèrent qu’ils avaient été introduits en contrebande à partir du Vietnam, à travers les zones frontalières non douanières de Dongxing. Ils ont ensuite été transportés par voie terrestre vers le Guangdong et d’autres lieux pour les livrer au receleur à des fins de vente. Trois suspects ont été arrêtés sur les lieux, 10 pangolins vivants [NDA, 11 les autres pangolins vivants ont été saisis sur le lieu de recel probablement], 5 pangolins congelés, 3 750 grammes d’écailles de pangolin, 4 aigles vivants, 9 pattes d’ours congelées et d’autres animaux protégés et leurs produits ont été saisis, le réseau de contrebande a été démantelé (source : Legal Daily-Legal Network, 27 mars 2019). À noter que d’autres saisies plus anciennes, recouvrant 19 pangolins, avaient été réalisées entre juillet 2017 et janvier 2018 par les douanes de Guangxi, province du sud de la Chine à la frontière du Vietnam.
Il y avait des pangolins adultes et adolescents. Leur état de santé était très dégradé. Leurs corps étaient recouverts d’éruptions cutanées et seize sont morts après des soins intensifs. L’autopsie de la plupart des pangolins morts a révélé que leurs poumons étaient enflés, contenaient un liquide écumeux et présentaient des symptômes de fibrose pulmonaire. Certains présentaient une hépatomégalie et une splénomégalie. 21 échantillons d’organes des poumons, du système lymphatique et de la rate, présentant des symptômes évidents, ont été prélevés chez 11 pangolins décédés pour découvrir les virus impliqués.

Vraisemblablement les pangolins ont été infectés par les trafiquants
L’étude méta-génomique des échantillons prélevés sur ces pangolins avait conduit Ping Liu et al. à publier en septembre 2019 la reconstruction de 68 séquences contiguës distinctes partageant des similarités avec de nombreux virus connus. Parmi eux se trouvaient des virus Sendaï et des coronavirus, dont la plupart étaient des coronavirus à SARS. Les auteurs de l’étude concluent que les pangolins, dont la majorité des échantillons étudiés contenaient le virus Sendaï d’origine humaine, étaient morts d’un syndrome respiratoire dû à ce virus.
Par la suite, en 2020, dans une autre étude approfondie, parue dans Plos Pathogène, ils ont publié la séquence extraite, la plus proche du SaRS-Cov2, appelée pangolin-CoV-2020 consensus. Cette séquence combine en fait 3 séquences quasi complètes (obtenues de 3 pangolins malades différents) avec plus de 99.5 % d’identité entre elles. Au niveau du génome complet, elle partage 90,3 % d’identité de séquence avec le SARS-Cov2 et 90,5 % avec le RaTG13. Le virus pangolin-CoV-2020, aussi appelé GD-pangolin-Cov, est donc autant éloigné des deux virus les plus proches de lui. Cependant, le RaTG13 est éloigné dans le temps de 7 ans par rapport au SARS-Cov2 alors que celui du pangolin que de 6 à 9 mois. Ce qui se reflète dans le fait que, de façon très intrigante, le domaine de liaison au récepteur ACE2 de la protéine S du virus du pangolin est à 96,8 % identique à celui du SARS-Cov2 (et partage 4 des 5 acides aminés clés pour la liaison) alors que celui du virus RaTG13, en théorie plus proche, n’est identique qu’à 89,6 %. Cela veut dire que le virus du pangolin était en fait plus adapté à l’homme du point de vue du pouvoir de pénétration cellulaire. Alors pourquoi son génome est-il plus éloigné globalement du SARS-Cov2 que celui du RaTG13 ? Nous nageons ici dans un paradoxe.
La conclusion générale des auteurs est que le pangolin est un hôte naturel pour les coronavirus et que le SARS-Cov2 est le résultat de recombinaisons multiples dans une grande variété de chauves-souris et d’autres animaux sauvages. Cela peut paraître infondé, car, au contraire de l’épidémie de 2003 où les anticorps spécifiques au SARS avaient été retrouvés dans un grand nombre de civettes d’élevage, on ne peut exclure que les pangolins aient été infectés par les contrebandiers chinois eux-mêmes. Les pangolins sont des animaux sauvages solitaires qui n’ont pas de contact avec les chauves-souris. Par contre, la logique nous dit que les contrebandiers, les receleurs et les vendeurs sur les marchés forment un microcosme où doivent tourner de nombreux virus, comme le virus Sendaï qui affecte également les rongeurs, ou bien également des coronavirus de chauve-souris puisqu’ils en font le trafic. Ce microcosme homme-animal sauvage de contrebande et autres animaux semi-sauvages de marchés, vivant en promiscuité plus ou moins insalubre, forme un résonateur d’évolution rapide par recombinaisons de virus. Les virus peuvent s’échanger constamment entre différentes espèces animales et l’homme. Il n’est pas inconcevable dans ces circonstances que des virus pré-pandémiques, non détectés, peuvent incuber pendant des mois et des années dans ce milieu. Les pangolins qui sont arrivés au centre de secours de Guangdong sont quasiment tous morts, soit directement dans les jours suivant leur arrivée (16 pangolins) soit quelque temps après, malgré des efforts considérables pour les sauver. Le quotidien chinois Southern Metropolis Daily titre le 20 avril 2019 : « Un autre vient de mourir. Il n’en reste plus que quatre en vie et ils ont un besoin urgent de l’aide d’experts ! »

Il est donc peu vraisemblable que tous ces pangolins sauvages aient pu si soudainement mourir en même temps d’un coronavirus endogène à leur espèce dont ils seraient une sorte de réservoir parallèle… La seule explication c’est qu’ils ont été contaminés par les contrebandiers et que le virus était tellement nouveau pour eux (de même que le Sendaï virus qui crée un syndrome respiratoire) qu’ils en sont morts. La prévalence des anticorps associés à des coronavirus à SARS chez les trafiquants d’animaux sauvages a été clairement établie en 2003 par le Centre de Contrôle des maladies du Guangdong. Des études séro-épidémiologiques faites à cette époque ont révélé que le SARS-CoV n’avait pas circulé significativement dans la population humaine avant 2002. Par contre, on trouvait des individus séropositifs avant cette période parmi des travailleurs des marchés d’animaux sauvages, documentant encore le risque de transmission à l’homme.
Il n’y a aucune raison que cette réalité ait pu changer depuis 2003, bien au contraire. Il s’agit là d’un phénomène bien connu des épidémiologistes. D’ailleurs, Ping Liu et al. sont obligés de le reconnaître à demi-mots lorsqu’ils rajoutent dans leur conclusion qu’il n’est pas clair si le coronavirus identifié est une « flore virale commune aux voies respiratoires des pangolins » et que « la genèse pathogénique de ce coronavirus chez les pangolins reste à élucider ». Nous sommes désolés de le dire, mais il s’agit là encore d’une contorsion sémantique destinée à ne pas admettre directement que des sous-lignées du SARS-Cov2, peu pathogènes, circulaient chez les contrebandiers chinois déjà au début de 2019.
L’hypothèse de recombinaisons de virus dans la région du RBD est imprécise et peu solide
Ping Liu continue ensuite ses « imprécisions » en affirmant que leur analyse de recombinaison montre que la protéine S du virus pangolin-CoV-2020 peut se reconstruire à partir de fragments de séquence du virus Bat-CoV-ZC45 ou Bat-CoV-ZXC21 et de fragments de RaTG13. Ce que Ping Liu et al. qualifient d’analyse de recombinaison n’est ni plus ni moins qu’une analyse visuelle à partir des alignements de séquences. Et malgré notre bonne volonté nous ne voyons qu’un seul endroit de recombinaison possible entre les 5 coronavirus. Il s’agirait d’une recombinaison entre le pangolin-Cov2-2002 et le RaTG13 au niveau du motif de liaison au récepteur ACE2 (RBM) dans le domaine de liaison (RBD).

Nous pensons qu’il n’est visiblement pas prouvé que par recombinaisons naturelles la protéine S du SARS-Cov2 puisse être reconstruite à partir de fragments des virus Bat-CoV-ZC45 ou Bat-CoV-ZXC21 et de fragments de RaTG13. Si un généticien veut répondre à France Soir sur ce point ou sur d’autres points que nous avons analysés dans ce chapitre, nous l’invitons à le faire.
Nous nous demandons comment une telle affirmation, non fondée, peut être publiée dans un journal du calibre de PLOS Pathogens. Elle peut s’expliquer par le désir des auteurs et de l’éditeur de prévenir la théorie complotiste d’une manipulation spécifique au niveau du RBD du virus RaTG13, ou d’un autre virus très voisin, avec échange de RBM. Les échanges de RBD entre coronavirus pour tester leur infectiosité ont été couramment pratiqués ces 10-15 dernières années, en particulier par Shi Zheng Li depuis 2007. La première expérience de ce genre remonterait à une vingtaine d’années à l’Université d’Utrecht aux Pays-Bas. Ces expériences démontrent que l’on peut ainsi échanger le tropisme d’espèce entre virus, c’est-à-dire leur faire franchir la barrière des espèces.
On peut se demander s’ils ne cherchent pas à devancer et prévenir toute idée que les virus séquencés par les chercheurs, affiliés à l’armée populaire chinoise, puissent avoir été utilisés dans des manipulations à gain de fonction. Nous ne pensons absolument pas cela, mais c’est dire le problème sous-jacent de crédibilité de la Chine. Notons à ce sujet, en a parte, que le gouvernement chinois subit une telle pression internationale sur la question de l’origine de l’épidémie qu’il ne peut s’empêcher d’utiliser les techniques de désinformation les plus grossières, telle que cette déclaration faite le 20 novembre par le porte-parole du ministre des affaires étrangères chinois sous-entendant que l’émergence du virus pouvait impliquer plusieurs nations. Cette déclaration tendancieuse fait suite à la parution dans le journal Tumori de patients cancéreux italiens positifs aux anticorps spécifiques du SARS-Cov2 dès septembre 2019 (voir partie 3 du chapitre).
Mais revenons aux recombinaisons virales. À l’évidence, la seule recombinaison possible au niveau de la protéine S, entre tous ces virus, qui a pu avoir lieu est celle entre le pangolin-Cov-2020 et le RaTG13. Cette information conforte l’idée que des virus précurseurs du SARS-Cov2 circulaient à bas bruit entre les hommes et les animaux dans le microcosme des marchés aux animaux sauvages. De quel coronavirus le RBM de la protéine S du pangolin provient-il ? Cela reste une question centrale à résoudre, car sur l’intégralité de sa longueur, soit 72 acides aminés (résidus 435 à 506), le RBM du virus des pangolins est 98,6 % identique au SARS-Cov2, avec une seule mutation H496Q (l’acide aminé histidine H échangé avec l’acide aminé glutamine Q à la position 496 critique pour la liaison). En fait, on serait tenté de dire qu’il est 100 % identique, car, sur le plan des interactions physico-chimiques impliquées, l’histidine et la glutamine sont très proches et souvent interchangeables. De ce fait, la mutation H496Q peut se produire par hasard sans que cela induise une modification réelle de l’affinité du RBM pour l’ACE2. Tout cela est d’autant plus étrange que le RBM du virus GD-pangolin-Cov à 14 % de différence avec le RBM du RaTG13 qui, par ailleurs, est un virus globalement plus proche du SARS-Cov2. Cela contredit les lois de l’évolution à tout point de vue. D’abord, comment le virus du pangolin pourrait-il être le résultat d’une recombinaison en 2018-2919 avec un virus de 2013 ? Ensuite, s’il était une recombinaison avec un virus descendant du RaTG13, ne devrait-il pas être plus proche du SARS-Cov2 que le RaTG13 sur l’ensemble de son génome ?
C’est ce paradoxe qui a conduit les généticiens à faire l’hypothèse quelque peu délicate que le RBM du pangolin aurait convergé par pression évolutive de façon optimale séparément en même temps dans le pangolin et l’homme. Une séquence de 72 acides aminés aurait donc convergé quasi -exactement et simultanément chez 2 hôtes aussi distincts que le pangolin et l’être humain. Cela est à notre avis extrêmement improbable, en tout cas considérablement moins probable que l’hypothèse scientifique alternative que les pangolins aient pu être infectés par l’homme avec un coronavirus apparenté au SARS-Cov2, d’une façon que nous ne comprenons pas (voir section suivante sur l’article d’Alina Chan et al.).

Pour finir, dans le registre contre toute idée d’une possible manipulation de virus, Liu et al. ajoutent également qu’un coronavirus RmYN02 à 93,3 % identique au SARS-Cov2 a été identifié à partir d’échantillons provenant de 227 chauves-souris capturées, entre mai et octobre 2019, au cours d’une énième campagne dans le Yunnan. Le virus RmYN02 a la particularité de présenter une insertion multiple à la jonction entre les sous-unités S1 et S2 de la protéine S, sans toutefois que cette insertion corresponde, comme dans le cas du SARS-Cov2, au site de clivage de la furine. La présence de ce site de clivage à un endroit qui renforcerait le pouvoir de pénétration du SARS-Cov2 est considéré par certains comme une preuve de manipulation. Nous étudierons ce problème au prochain chapitre.
Le mystère du RBM du GD-pangolin-Cov est renforcé par le GX-pangolin-Cov
Une seconde analyse par Tsan-Yuk Lam et al. a eu lieu également entre janvier et février 2020 sur 25 échantillons provenant de 18 autres pangolins saisis par les douanes de Guangxi. Il n’est pas précisé l’état de santé de ces pangolins lors de leur saisie, mais il est probable qu’ils aient été également transférés dans un centre de protection de la faune sauvage. Cependant, malgré une durée de vie pouvant atteindre 13 ans, ces pangolins sont tous morts et les échantillons ont été conservés par l’Université de Guangxi avant d’être envoyés pour analyse, en janvier 2020, à Tsan-Yuk Lam et al. Ces derniers ont extrait au total 6 génomes quasi complets de coronavirus qui partageaient entre 85.5 % et 92.4 % de similarité de séquences avec le SARS-CoV-2. Le génome le plus proche a donné le jour à un second coronavirus consensus appelé le GX-pangolin-Cov. Il se retrouve logiquement être le 3e coronavirus le plus proche du SARS-Cov2, derrière le RaTG13 et pangolin-Cov-2020 (GD-pangolin-Cov) avec 84 % d’identité de séquence avec le SARS-Cov2.
Lam et al. ne précise pas l’identité de séquence globale entre les virus extraits des pangolins saisis en 2017 et ceux saisis en 2019, mais elle se trouve autour de 94 %. Cela est gênant par ce qu’à 15 mois d’intervalle un virus endogène au pangolin n’aurait pas pu changer autant. La composition en acides aminés (aa) du domaine de liaison au récepteur ACE2 est à 89,2 % identique (21 aa diffèrent sur les 194 que compte le RBD) entre ces deux virus. Mais 86 % des mutations (18) se situent au niveau des 72 aa de la partie du domaine impliqué directement dans la liaison (RBM). Autre fait de première importance, l’analyse des mutations dans le RBM montre que 9 des 10 aa de contact avec l’ACE2, sans être critique pour la liaison avec l’ACE2, restent identiques entre les 2 virus. Par contre, 4 des 5 aa qui jouent un rôle critique dans la liaison ne correspondent pas. Cela est tout à fait étonnant et indiquerait que le RBM aurait muté en 15 mois chez les pangolins pour résulter dans un RBM 98,6 % identique à celui du SARS-Cov2 humain. Cela implique soit un taux de mutation non synonyme sur cette section du génome tout à fait hallucinant (impossible à obtenir en l’absence d’un forçage comme celui décrit au chapitre 2) soit une recombinaison aléatoire favorable du RBM tout à fait exceptionnelle puisqu’elle correspondrait à un virus quasi optimal pour l’être humain.

Partie 5 — Le SARS-Cov2 était déjà adapté optimalement à l’homme dès son apparition fin 2019
Alina Chan et al. ont montré que sur 151 isolats de virus collectés (50 par mois de janvier à mars 2020) il ne s’était produit que 2 mutations du RBM (sur des aa ni de contact ni critiques pour sa liaison avec l’ACE2) concernant 2 isolats distincts. Un point jamais soulevé est que le pangolin n’est certainement pas le mammifère possédant le récepteur ACE2 le plus proche de l’homme. Ce qui rend quasiment improbable, si le pangolin était porteur naturel de ce virus, qu’en passant à l’homme le virus ne se soit pas adapté davantage au niveau du RBD ou du RBM de la protéine S.
Donc, sur ce point nous rejoignons Alina Chan et nous analysons que l’hypothèse retenue généralement, d’une recombinaison multiple au niveau du RBD à travers plusieurs espèces animales, n’est pas fondée sur des preuves concrètes. Il ne s’agit que de spéculations non conclusives. Qu’une telle recombinaison dans des animaux très distincts de l’être humain ait pu converger vers un motif de liaison RBM du RBD quasi 100 % adapté à l’homme par hasard est très peu probable, les virus s’adaptant naturellement à leur hôte. Par contre, les récepteurs ACE2 de 18 primates (chimpanzé, orang-outan…) sont 100 % identiques à celui de l’homme sur les 25 aa directement impliqués dans la liaison avec la protéine S. Ce fait publié par l’Académie des Science Américaine (PNAS) est tenu totalement à l’écart des débats. Cela est si dérangeant que l’article dans le journal PNAS prévient laconiquement dans une section, intitulée « Signification » placée en tête de l’article, qu’il ne faut pas sur-interpréter cette donnée du problème dans la recherche de l’hôte intermédiaire. Par exemple, le rhinopithèque de Roxellane, qui appartient au groupe des 18 primates dont le motif de liaison de l’ACE est identique à celui de l’homme, est une espèce en voie de disparition qui se trouve dans les forêts des régions montagneuses des provinces du centre de la Chine (Gansu, Shaanxi et Hubei). Ces animaux vivants dans les arbres peuvent être en contact occasionnel avec des chauves-souris ou leurs excréments. Ils pourraient donc avoir joué le rôle d’hôte d’intermédiaire plus sûrement que les pangolins.
À Wuhan des primates de différentes espèces peuvent se trouver dans les zoos certainement, sur les marchés aux animaux très possiblement, et surtout à l’Institut de virologie de Wuhan. Les singes de laboratoire sont très courants dans les laboratoires de virologie, car ils servent a mettre en pratique et tester les hypothèses au cours de l’élaboration des vaccins. On peut donc imaginer le scénario d’un virus expérimental, qui serait passé d’un singe de laboratoire à un technicien ou un employé s’occupant d’eux, puis aurait gagné le marché aux animaux de Huanan où un des marchés annexes, où des animaux et d’autres êtres humains auraient été contaminés.
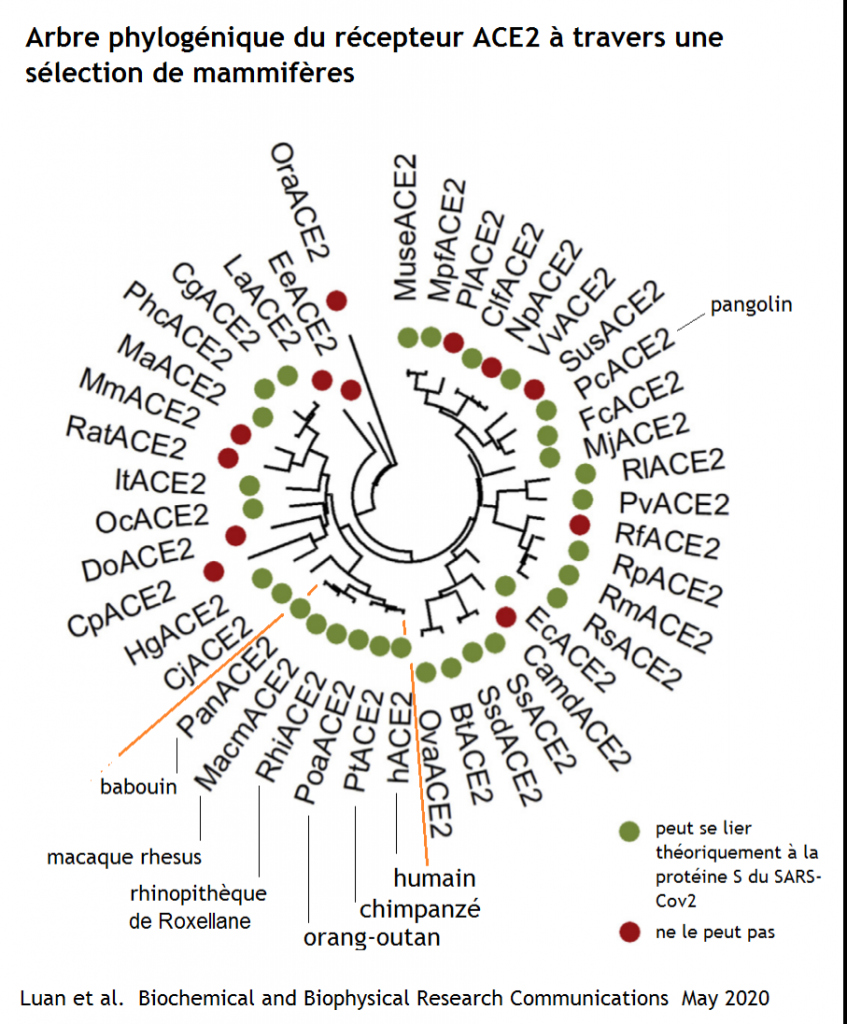
Dans le cas d’une recombinaison, quel serait cet autre virus avec un RBM déjà optimal pour infecter l’être humain ? Proviendrait-il d’un autre hôte intermédiaire dont le récepteur serait également très proche, ou bien de chauves-souris qui selon la théorie de Shi Zheng Li véhiculent des coronavirus possédant des protéines S qui permettent d’infecter l’homme, directement sans adaptation.
Lam et al. concluent leurs travaux de façon, nous semble-t-il, plus rationnelle que Liu et al. Ils disent en substance : « Cela suggère que ces animaux sont un hôte important de ces virus, ce qui est surprenant étant donné que les pangolins sont des animaux solitaires avec des effectifs de population relativement faibles, reflétant le statut d’espèce en danger. En effet, sur la base des données du moment il ne peut être exclu que les pangolins aient acquis leur virus apparenté au SARS-CoV-2 indépendamment des chauves-souris ou d’un autre animal hôte. » Cela ouvre implicitement la porte à une contamination humaine.
Comment Alina Chan montre que le SARS-Cov2 était optimalement adapté à l’homme dès son apparition fin 2019
Alina Chan et son équipe ont comparé la dynamique évolutive du SRAS-CoV et du SARS — CoV2. À cet effet, ils ont utilisé pour le SARS-Cov 11 génomes de la phase épidémie précoce à moyenne et 32 génomes de la phase épidémique tardive. Ils ont également utilisé 46 génomes de SRAS-CoV-2 dont un isolat de Wuhan, du début décembre 2019, et 15 génomes provenant de diverses régions géographiques, recueillis au hasard chaque mois de janvier à mars 2020.
Ils ont constaté que la diversité génétique du SRAS-CoV-2 était bien plus faible que celle du SRAS-CoV, dont la diversité était considérable dans la phase précoce et moyenne de l’épidémie. La pression sélective avait été la plus élevée lors du passage du virus des civettes palmistes aux humains et avait diminué vers la fin de l’épidémie. Cette série d’adaptations entre espèces animales et l’homme a abouti à un SRAS-CoV hautement infectieux qui a dominé la phase épidémique tardive.

En comparaison, le SRAS-CoV-2 présente une diversité génétique plus similaire à celle du SRAS-CoV de la phase épidémique tardive. Le gène de pointe S, et les Orf3a et Orf1a avaient subi une forte pression sélective dans l’épidémie de SRAS-CoV. Les taux de mutations synonymes (dS) et non synonymes (dN) des gènes de la protéine pointe S, de l’Orf3a et l’Orf1a du SRAS-CoV-2 sont similaires à ceux de l’épidémie tardive de SRAS-CoV. En revanche, les quelques rares substitutions non synonymes dans le gène de pointe S du SRAS-CoV-2 n’a pas conféré d’avantage adaptatif dans l’intervalle de temps étudié.
Alina Chan et ses collaborateurs en concluent qu’au moment où le SRAS-CoV-2 a été détecté pour la première fois à la fin de 2019, il était donc déjà complètement adapté à la transmission humaine. Leurs observations semblent indiquer une phase d’adaptation invisible du SARS-CoV-2. Cela pourrait s’expliquer par le fait que le SARS-Cov2 est beaucoup moins létal que le SARS-Cov, avec de très nombreux cas asymptomatiques et pauci symptomatiques qui a pu favoriser sa circulation pré-pandémique non détectée. Pendant cette période le virus a pu accumuler des mutations adaptatives qui ont renforcé sa contagiosité et sa pathogénie à l’automne 2019.
Seuls 4 échantillons environnementaux provenant des prélèvements effectués au marché de Huanan ont permis d’obtenir des génomes complets de virus. Quant aux séquençages des quelques échantillons animaux mis à disposition des chercheurs par le CDC chinois, ils sont fortement contaminés et inexploitables. Les 4 génomes qui ont pu être résolus partagent plus de 99,9 % d’identité avec l’isolat Wuhan Hu-1 (premier virus isolé d’un patient de décembre). Une identité > 99,9 % n’avait été observée en 2003-2004 qu’entre des isolats rapprochés dans le temps provenant d’individus d’une même espèce, soit l’être humain soit les civettes, mais pas entre humains et civettes. L’identité de séquence la plus élevée enregistrée entre humains et civettes avait été de 99,78 %. Alina Chan conclue que les échantillons du marché de Huanan ont donc été contaminés par des vendeurs ou des clients, mais pas par des animaux intermédiaires.
Il est important de rappeler qu’il y a eu deux épisodes de SRAS-CoV en 2002-2004, chacun résultant de transmission de la civette palmiste à l’homme : le premier est apparu à la fin de 2002 et s’est terminé en août 2003 ; le second est né à la fin de 2003 à partir d’une population persistante de progéniteurs du SRAS-CoV chez les civettes. La deuxième épidémie a été rapidement réprimée grâce au suivi diligent des hôtes humains et animaux, retenant les leçons de la première épidémie. Pour prévenir aujourd’hui des flambées consécutives similaires de SRAS-CoV-2, il est essentiel de tirer les leçons du passé et de mettre en œuvre des mesures pour minimiser le risque que d’autres précurseurs de type SRAS-CoV-2 s’adaptent et réapparaissent parmi les humains.
Alina Chan estime qu’il serait étrange si aucun précurseur ou proche parent du SARS-CoV-2 n’était pas découvert chez l’être humain ou chez les animaux. Elle suggère d’évaluer la prévalence de certains anticorps potentiellement reliés au SARS-Cov2 chez les vendeurs et les trafiquants des marchés, pour déterminer si des précurseurs proches du SARS-CoV-2 ont pu circuler dans cette communauté. Elle propose de prélever des échantillons d’animaux sauvages et d’élevage ainsi que des échantillons humains déposés dans des banques de données médicales avant le départ de l’épidémie.
Elle dit également que séquencer plus d’isolats précoces du virus de Wuhan permettrait d’identifier des virus progéniteurs proches moins bien adaptés à l’homme, comme ça a été le cas pour l’épidémie de SARS de 2003. Elle conclut en ajoutant que pendant que ces recherches s’effectueraient, il serait prudent de limiter l’activité humaine qui conduit à de fréquents et prolongés contacts avec des animaux sauvages et leur habitat. Cela nous paraît frapper au coin du bon sens, mais il est évident que la Chine n’a aucune intention ni de lever le voile sur l’origine de l’épidémie ni d’interdire rigoureusement le trafic d’animaux sauvages.
Pour finir elle n’exclut aucun scénario, que ce soit : (a) la possibilité d’un intermédiaire animal autre que le pangolin ; (b) l’adaptation interhumaine du virus ; (c) un accident de laboratoire, et préconise de prendre des mesures pour prévenir chacun de ces scénarios dans le futur.
Conclusion
Le récepteur ACE2 du pangolin n’est pas le plus proche de celui de l’homme et l’adaptation parfaite du RBM du virus GD-pangolin-Cov reste un mystère. Si le pangolin avait été l’hôte intermédiaire, les virus extraits devraient avoir 98 ou 99 % d’identité de séquence avec le SARS-Cov2. Il est quasi certain, pour ne pas dire certain, que les pangolins de contrebande ne sont que de malheureuses victimes, dont l’autopsie post mortem nous livre des informations capitales sur le timing de la genèse de l’épidémie. Entre août 2017 et janvier 2018, des coronavirus à SARS circulaient chez les trafiquants d’animaux sauvages en relation avec les receleurs des marchés et les vendeurs. D’ailleurs, il est très probable que les trafiquants étaient immunisés du fait de leur promiscuité avec toutes sortes d’hôtes intermédiaires potentiels. N’oublions pas également que la létalité du SARS-Cov2 n’a rien à voir avec celle du SARS-Cov. En mars 2019, sont apparus probablement dans ce microcosme des coronavirus à SARS optimaux sur le plan de la liaison au récepteur ACE2. À l’exception d’une mutation faiblement impactante H496Q à une position critique, l’adaptation complète du RBM s’est donc produite entre janvier 2018 et mars 2019 et a pu impliquer des contagions croisées multiples entre des animaux sur des marchés et des hommes. Mais cela n’exclut pas un accident de laboratoire non loin d’un marché géant… En tout cas, l’année 2018 est bel et bien une date clé de l’histoire du Covid-19. Une chose est certaine, cette possibilité ne pourra jamais être vraiment refermée en raison de la destruction des échantillons d’animaux saisis sur le marché de Huanan, l’absence d’enquête officielle sur les premiers patients atteints et le refus de la Chine de laisser une enquête internationale se dérouter à l’Institut de virologie de Wuhan.
Mais beaucoup de gens ont intérêt à ce que l’attention internationale soit détournée du laboratoire P4 de Wuhan, dont l’Institut Pasteur et d’autres instances scientifiques qui contrôlent la science mondiale comme le Journal Nature. Dans un article de Nature du 23 novembre, on apprend que des échantillons de chauves-souris, conservés depuis 2010 à l’Institut Pasteur de Phnom Penh au Cambodge et depuis 2013 au Japon, contiennent des coronavirus du type SARS. La belle affaire, le virus extrait de l’échantillon japonais à 81 % d’identité de séquence avec le SARS-Cov2, c’est-à-dire bien inférieur au 88 % des virus isolés par les recherches militaires chinoises, et le pourcentage d’identité n’est pas révélé pour celui de l’échantillon cambodgien, alors que non venons de montrer qu’il ne faut qu’une dizaine de jours seulement pour séquencer un virus. Si ce virus est un ancêtre direct du SARS-Cov2 il y a 10 ans, alors il sera dans la zone des 93-96 % d’identité selon les calculs que nous avons présentés ici. Mais alors, quid des 8 autres coronavirus de type SARS prélevés dans la mine de Mojiang ?
Dans le prochain chapitre, nous examinerons minutieusement les anomalies du génome du SARS-Cov2, comme la présence d’un site clivage de la furine qui semble renforcer son pouvoir de pénétration, et les travaux du Professeur Luc Montagnier et du mathématicien Jean-Claude Perez, sur leur hypothèse d’insertion d’épitopes du virus du SIDA et de l’agent infectieux du paludisme.
chapitre 7
Partie 1 — Manipulation des virus par enzyme de restriction et PCR
Où l’on apprend que l’on sait depuis 2002 manipuler le génome d’un coronavirus sans laisser de trace et que l’Institut Pasteur ne dit pas tout sur ses brevets et le reste.
Sans que la majorité d’entre nous en fût consciente, le prix Nobel de chimie de 2020 nous a projetés officiellement dans un monde de science-fiction. Cependant, la manipulation d’un génome complet de coronavirus était déjà possible depuis l’année 2002, grâce à des techniques simples et performantes dont certaines permettent de ne pas laisser de trace.
Première création d’un coronavirus chimérique synthétique en 2000
Il y a tout juste 20 ans, Lili Kuo, affiliée au David Axelrod Institute – New York State Department of Health, en collaboration avec le groupe de recherche de J.M. Rottier à l’Université d’Utrecht (Pays-Bas), publiait le résultat de travaux séminaux qui allaient devenir, dans les années suivantes, la base conceptuelle des recherches que Shi Zheng Li a poursuivi sur la transmissibilité des coronavirus de chauves-souris à l’homme. Le titre de l’article décrivant cette première mondiale en termes de manipulation de virus laisse peu de place au doute : « Redéfinir l’hôte cible d’un coronavirus par échange de l’ectodomaine de sa protéine S : franchissement de la barrière cellulaire des espèces hôtes. »
Ils avaient construit un mutant du coronavirus de l’hépatite murine (MHV) dans lequel une partie de la protéine S (ectodomaine ou sous-unité S1 située à l’extérieur de la membrane du virus) avait été remplacée par le domaine équivalent, mais très dissimilaire (84 % de divergence) de la protéine S du coronavirus de la péritonite infectieuse du félin (PIF du chat). Il faut noter que ces 2 coronavirus sont hautement spécifiques de leurs hôtes naturels, empêchant la contamination entre les deux espèces. Le virus chimérique artificiel obtenu (fMHV) avait acquis la capacité d’infecter les cultures cellulaires félines et en même temps avait perdu celle d’infecter les cultures cellulaires de tissus murins. Ces chercheurs avaient ainsi démontré, par échange réciproque de facteurs moléculaires infectieux, le concept que la spécificité des coronavirus pour un hôte particulier était déterminée par l’interaction de la protéine S avec le récepteur cellulaire de l’hôte. De plus, ils avaient identifié la région responsable de cette spécificité comme étant une séquence d’environ 70 acides aminés située à l’extrémité C-terminale de la sous-unité S1, appelée motif de liaison au récepteur (RBM, voir chapitre 6, partie 4). Ces résultats sont fondateurs de ce qui s’est ensuite passé depuis 20 ans. Il est important de mentionner ici que les auteurs avaient noté cependant, que le virus chimérique recombinant fMHV semblait être moins infectieux que le virus souche naturel MHV.
Ils ont attribué ce « problème », entre autres, à la possibilité que le système de transcription du virus murin récepteur de la manipulation ne soit pas adapté de façon optimale à la réplication de la protéine S féline chimérique. Cela aurait engendré un nombre moins élevé de copies du gène S au cours de la réplication et donc un pouvoir infectieux diminué. Une autre hypothèse était que la protéine S hybride construite ne s’intégrait pas aussi bien dans la membrane du virus murin que la protéine S murine, déstabilisant aussi les capacités infectieuses du virus mutant. Cet aspect des choses est très important, car il montre que l’on ne peut pas réellement fabriquer un organisme optimal de novo, directement par assemblage d’éléments distincts, même provenant de la même espèce.
À une autre échelle, ce phénomène est comparable à celui de rejet dans les greffes ou les transfusions sanguines. Pour qu’un fonctionnement soit optimal, il faut une adaptation parfaite que seule la pression évolutive peut engendrer. Malheureusement, dans le cas des virus on peut par passages répétés, dans dans une culture cellulaire ou chez un mammifère hôte (voir chapitre 2), forcer l’adaptation. D’ailleurs, Kuo et al. décrivent comment d’autres études ont réussi à forcer le passage interespèces du virus MHV de cette manière, le virus gardant la capacité d’infecter les souris. Ce dernier point est à notre avis crucial dans la compréhension de la genèse du Covid-19, car il montre qu’une exposition répétée à un coronavirus spécifique d’une espèce (les chauves-souris) permet le franchissement de la barrière interespèce sans que le virus ne perde sa capacité par rapport à l’espèce initiale. Donc, l’exposition répétée des trafiquants des marchés avec des animaux sauvages comme des chauves-souris, et l’exposition des animaux entre eux, est bel et bien un mécanisme de franchissement des espèces avec acquisition de transmission interhumaine, inter-animale et humaine-animale qui permet ensuite, la circulation et la recombinaison de nombreuses souches de coronavirus. Ce mécanisme est plus lent dans les marchés que dans des cultures cellulaires à haute concentration. Mais le principe est le même, une promiscuité permanente lui permet d’avoir lieu sur quelques mois ou quelques années au plus.
Avant 2012, on manipulait un génome par recombinaison ciblée d’ARN
La manipulation de virus consiste à en faire la rétro-ingénierie, c’est-à-dire à réassembler des éléments fractionnés, dont certains ont été modifiés à dessein, par génie génétique. Une difficulté supplémentaire avec les virus à ARN est que certaines manipulations doivent passer par l’intermédiaire d’un ADN complémentaire (ADNc), dit recombinant, codant le génome du virus alors que le virus à ARN ne possède jamais de code ADN à son état naturel. À l’époque de Kuo et al., les procédés de microbiologies utilisés étaient très complexes dans leur conception et également dans leur mise en œuvre, ce qui nécessitait de longs efforts s’étalant sur des années. Les expériences de génie génétique publiées par de Kuo et al. en 2000 se sont déroulées sur une période de 2 ans, suivant une phase d’élaboration en 1997 d’un premier virus génétiquement préparé pour permettre les expériences de recombinaison d’ARN ciblant la protéine S des virus félin et murin.
Ils ont utilisé la mutagenèse dirigée qui fait appel à l’utilisation d’un plasmide dans lequel on insère artificiellement une séquence modifiée d’un gène ou d’une partie d’un gène. Les plasmides sont de l’ADN circulaire (pouvant dépasser une longueur de 50 kb) présent naturellement dans les bactéries. Leur rôle naturel est de conférer aux bactéries un pouvoir de défense, car ils encodent des enzymes permettant la lyse des antibiotiques. Les plasmides sont au cœur des procédés de génie génétique. Réinjectés après manipulations dans les bactéries ils permettent l’expression de protéines exogènes manipulées, facilement et en grande quantité, engendrant de multiples applications. Mais le champ d’application le plus critique qui a été ouvert était la possibilité de manipuler des virus.
Cependant, en 2000, la taille très grande des génomes des coronavirus, environ 30 000 nucléotides (nt), les rendait inappropriés pour des manipulations du type mutagenèse dirigée. Les nucléotides sont 4 bases azotées : A, G, C, T dans le cas de l’ADN et A, G, C, U dans le cas de l’ARN, qui sont les briques élémentaires constituant les génomes (voir chapitre 5). Kuo et al. ont contourné le problème en utilisant la propriété naturelle de recombinaison des coronavirus. Ils ont exprimé dans une culture de bactéries un virus MHV muté (Alb4), qui avait perdu sont gène N (NDA, l’absence de la protéine N empêche la nucléocapside de se former et expose l’ARN du virus au cytoplasme bactérien ce qui permet les réarrangements), en même temps qu’un vecteur donneur de l’ARN de la protéine S féline plus la partie restante du virus jusqu’à l’extrémité 3′ avec le gène N intègre. Le processus de recombinaison a généré aléatoirement des virus mutants qui ont pu être séparés ensuite entre ceux actifs sur les cellules murines et ceux actifs sur les cellules félines. Ensuite, on pouvait vérifier que ceux actifs sur les cellules félines ne l’étaient pas sur les cellules murines.
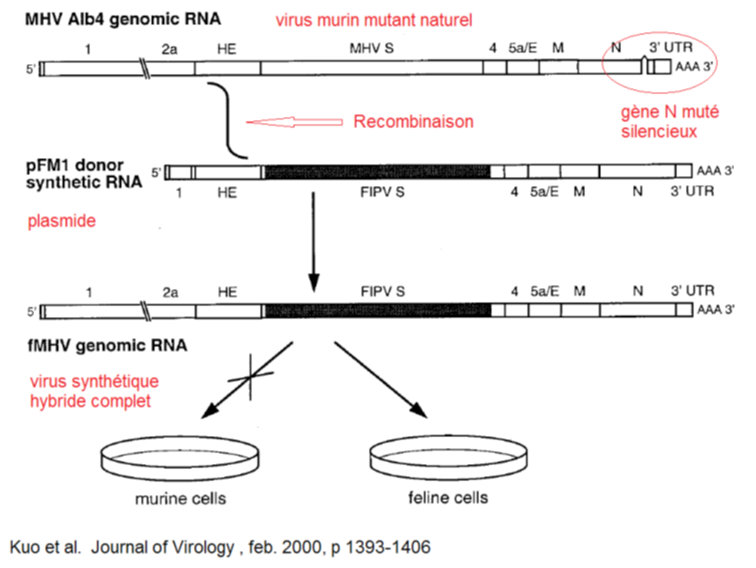
Dès le début des années 1980, on savait faire la manipulation moléculaire des virus à ADN en passant par le clonage moléculaire d’une partie ou de la totalité du génome viral dans un plasmide bactérien. Une difficulté pour les virus à ARN provenait du fait que l’ARN ne peut pas être cloné directement dans un plasmide qui ne peut contenir que de l’ADN. Cet obstacle a commencé à être levé à partir de 1978 avec le virus bactériophage Qβ. En 1981, le clonage d’un ADNc de l’ARN du poliovirus a été effectué dans un plasmide bactérien. L’ARN du virus a été transcrit en ADNc par une transcriptase inverse et puis incorporé dans le plasmide bactérien, par l’intermédiaire d’enzymes de restriction et de ligases qui coupent le plasmide et le recollent ensuite quand l’ADNc s’y est introduit. L’introduction de l’ADNc avait été réalisée grâce à une modification chimiquement préalable, à chacune de ses extrémités, pour y introduire un site de clivage identique (d’une longueur de quelques nucléotides) à celui permettant d’ouvrir le plasmide. En effet, des petites séquences d’ADN contenant un ou plusieurs sites de clivage peuvent être synthétisées chimiquement à volonté. Ensuite, en faisant agir une enzyme de restriction spécifique sur l’ADNc, cela a permis de créer des coupures complémentaires avec des côtés spécifiques à ceux engendrés par la coupure dans le plasmide. Les extrémités de l’ADNc ayant donc une affinité pour les extrémités complémentaires de la coupure se sont assemblées naturellement et ont refermé le plasmide en s’y insérant. La finalisation des jointures a été assurée par une ligase. Ce résumé schématique de l’expérience ne rend pas compte d’un grand nombre de difficultés techniques nécessitant de multiples étapes dans la construction et l’insertion de l’ADNc dans le plasmide. Elles sont dues à la difficulté d’utiliser les enzymes de restriction correctement et en particulier obtenir l’insertion dans la bonne orientation. À noter qu’à présent, pour obtenir la bonne orientation du premier coup on fait appel à 2 sites de restrictions distincts à chaque extrémité de l’ADNc et on coupe le plasmide en 2 endroits portant les mêmes sites de restriction. Les plasmides synthétiques modernes intègrent des foultitudes de sites de restriction.
Le plasmide, augmenté de l’ADNc du virus, a été alors injecté (transfection) dans des cultures de cellules humaines et de primates et sa transcription par l’ARN-polymérase cellulaire pouvait engendrer un ARN messager viral complet capable d’engager un cycle productif dans les cellules transfectées. La voie était ainsi ouverte pour la manipulation de n’importe quel virus à ARN. Depuis, les techniques se sont considérablement améliorées.
Avec les plasmides, les enzymes de restriction sont un outil incontournable essentiel dans les manipulations génétiques. Elles sont produites naturellement par des bactéries comme mécanisme de défense contre les infections par les bactériophages, des virus spécifiques des bactéries. Lorsque le virus injecte son ADN dans la bactérie, celui-ci est coupé par l’enzyme de restriction au niveau de ses sites spécifiques d’une longueur de 4 à 10 paires de bases. Il en existe un grand nombre répertorié, toutes spécifiques à une séquence particulière. Le hasard fait que l’on peut retrouver dans tous les génomes jusqu’aux organismes évolués, comme les mammifères, des sites de clivages répartis aléatoirement, ouvrant la possibilité de manipulation d’insertion ou d’échange de séquences ADN.
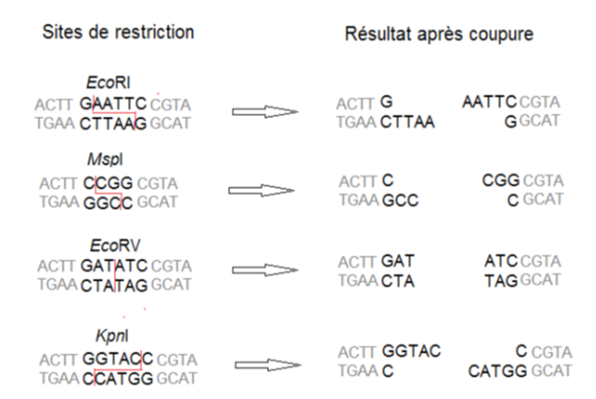
Cependant, leur répartition est aléatoire et suivant le nombre de nucléotides reconnus, les enzymes de restriction coupent l’ADN plus ou moins fréquemment. Par exemple, les enzymes de restriction MspI (CCGG) et BamHI (GGATCC) coupent en moyenne l’ADN toutes les 256 (44) et 4096 (46) paires de bases. Cela restreint considérablement les possibilités d’action ciblée à volonté, car on s’expose à couper l’ADN cloné en plusieurs endroits au lieu d’un seul. Cependant, la technique PCR permet de corriger ces problèmes en mutant les sites de restrictions indésirables et en restaurant la séquence originale par la suite par manipulation inverse. Des manipulations par un autre outil beaucoup plus complexe à mettre en œuvre à base de protéines à doigt de zinc peuvent également avoir lieu. Ces protéines reconnaissent spécifiquement des codons (groupe de 3 paires de bases) et il y a en existe pour toutes les possibilités. Une fois assemblée et couplées à d’autres enzymes dans un complexe supramoléculaire on peut arriver à manipuler un endroit du génome. Mais la mise en œuvre en termes de temps et de savoir-faire technique est plus que décuplée.
Par contre, des mutations ponctuelles peuvent être introduites très facilement depuis les années 1990 dans des génomes viraux par la technique PCR (réaction en chaîne par polymérase), une technique dont les Français ont découvert à leur détriment l’existence au cours de l’épidémie de SARS-Cov2. Cette technique très souple d’utilisation permet par exemple, comme nous l’avons dit dans le paragraphe précédent, de protéger un site de restriction à un endroit du génome pour empêcher son clivage.

Nous devons ici ouvrir une parenthèse indispensable pour l’information du public. La façon dont la PCR est utilisée dans les tests mis en place par les autorités de santé permet de repérer la présence d’une séquence complémentaire d’une séquence amorce d’une longueur très réduite (20 à 30 kb) par rapport à la longueur d’un génome intégral de virus. Elle ne détecte pas la présence de virus, mais seulement de fragments dénaturés de virions. Ces fragments sont présents dans les fosses nasales au cours de l’infection, mais également longtemps après de sorte que très nombreuses personnes ayant développé le virus de façon asymptomatique, ou pauci symptomatique, se retrouvent comptabilisées à tort comme nouveaux cas d’infection. De plus, les polymérases engendrent naturellement des erreurs de copies de sorte qu’au-delà de 30 cycles de réplication le test va être faussement positif dans un certain nombre de cas. À 40 ou 45 cycles de réplication (comme il a été pratiqué en France en octobre et probablement jusqu’à mi-novembre 2020) le nombre d’erreurs explose de façon exponentielle, engendrant un nombre considérable de faux positifs. L’étude universitaire de Rita Jafaar et al à l’IHU Méditerranée, où la présence virale a été vérifiée par culture cellulaire des échantillons utilisés pour la PCR, montre qu’à 35 cycles seulement 3 % des tests PCR positifs contiennent effectivement un virus viable. Cela se traduit par un taux de tests PCR faux positifs de 97 % ! Nous sommes donc là en présence d’une manipulation dictatoriale du peuple français que l’on a piégé de façon éhontée pour lui arracher son consentement de privation anticonstitutionnelle de liberté.
Mais revenons à nos moutons, les plasmides contiennent naturellement des sites de clivage d’enzymes de restriction dont ils sont protégés par le mécanisme général de méthylation. Des plasmides génétiquement modifiés existent qui comprennent, entre autres, de nombreux sites de clivages qui permettent des manipulations à volonté, mais également des protéines fluorescentes qui servent de marqueurs au cours des expériences pour vérifier que le plasmide a bien été intégré (transfection) par les cellules où le virus doit être exprimé. De plus des gènes de résistance aux antibiotiques présents également dans le plasmide permettent d’éliminer les cellules « non-transfectées » par ajout d’antibiotique.
Il est possible depuis 2002 de manipuler le génome complet d’un coronavirus de type SARS sans laisser de trace
Il existe des familles de plasmides qui permettent le clonage et la fusion de gènes sans laisser de trace (seamless cloning). Cette technique, qui existe depuis le début des années 2000 est appelée avec humour « No see’m », c’est-à-dire « Vous ne les voyez pas » (sous entendu les sites de restriction). Elle permet l’assemblage facile de protéines hybrides avec précision sans que des petits morceaux de séquences non désirés soient introduits. Yount et al. ont rapporté dès 2002 le clonage complet du coronavirus de l’hépatite murine (d’une longueur 31 kb) par intégration de son ADNc recombinant (c’est-à-dire reconstruit à partir de fragments) dans un plasmide de ce type. Les jonctions des sites de restrictions qui étaient situées à la fin de chaque fragment d’ADNc avaient été systématiquement éliminées (par enzyme de digestion) pendant l’assemblage du génome complet, permettant d’obtenir un clone du virus sans introduire le moindre changement au niveau de la séquence de nucléotides. Le virus ainsi cloné avait gardé toute sa capacité d’infectiosité. Cette approche avait permis d’éviter les changements au niveau de la séquence qui sont normalement associés avec la construction de l’ADNc complet d’un génome viral.
Cette méthode très puissante fait appel à des enzymes de restriction de type IIS et des sites de restriction spéciaux (Esp3I) qui disparaissent ensuite au cours de la fusion des fragments. Par exemple, en la combinant avec la PCR, il est possible d’insérer des sites Esp3I à n’importe quelle position dans un génome viral et créer un domaine variable susceptible d’être modifié à volonté par mutagenèse ciblée, par PCR ou tout simplement échangé avec une autre séquence, sans qu’il y ait la moindre preuve que des sites de restriction ont été utilisés dans les manipulations d’ADN recombinant. Au final on peut vérifier également par PCR que le virus a été exprimé correctement dans les cellules transfectées et le récolter pour le tester sur d’autres cultures cellulaires, par exemple sur des tissus de mammifères (tissus pulmonaires, etc.) voire être directement testés sur des animaux vivants comme des souris, des furets ou des primates de laboratoire.
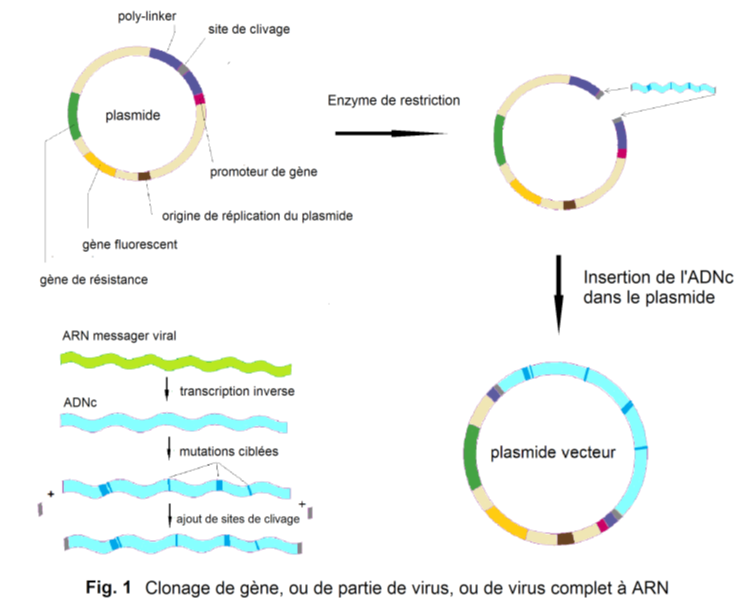
Finalement, il faut comprendre qu’une mutation ponctuelle dirigée effectuée par PCR sur un virus entièrement cloné sans modification de séquence dans un plasmide ne laisserait pas de trace.
On peut également échanger à l’intérieur d’un gène une séquence complète d’ADN sans laisser de traces à condition qu’il existe naturellement dans le génome du virus, au préalable, des sites de clivage par enzymes de restriction en amont et en aval de la séquence que l’on désire manipuler. Cela arrive fréquemment (voir partie 3 du chapitre), mais pas absolument n’importe où. Par contre, il est parfaitement possible d’introduire artificiellement par PCR un site de clivage dans une zone inter-gène ou même à l’intérieur d’un gène en repérant habilement des endroits où des mutations synonymes le permettent, de sorte à préserver l’intégrité de la composition en acides aminés. Mais cela n’est pas absolument nécessaire non plus, car, une fois la manipulation effectuée les sites de clivage peuvent de toute façon être effacés par mutation ponctuelle dirigée par PCR de la même façon qu’ils ont été introduits. Évidemment, tout cela demande un travail supplémentaire, mais reste très possible dans un temps raisonnable. Quant aux sites de restriction qui relient l’ADNc au plasmide, ils sont situés avant le codon d’initiation de la traduction (AUG) à l’extrémité 5′ et après le codon stop (UAA, UAG, ou UGA) à l’extrémité 3′ de sorte qu’ils n’apparaissent pas dans le produit génétique final.
L’insertion artificielle de sites de clivages est nécessaire lorsque l’on veut substituer une fraction de séquence avec une autre entièrement synthétique parce qu’on est limité par une longueur n’excédant pas 100 à 200 nucléotides. Le rendement et le taux d’erreurs générées par la synthèse chimique nucléotide par nucléotide ne permettent pas d’aller au-delà. Il faut donc introduire, par une série de mutations ponctuelles, effectuées par PCR, des sites de restriction encadrant très précisément la région de la séquence cible des manipulations. Par exemple, dans les travaux sur le SARS-Cov les chercheurs de l’Institut de virologie de Wuhan ont artificiellement inséré des sites de restrictions autour du motif de liaison (RBM, long de 213 nt) au récepteur ACE2 pour l’étudier. Comme le pointe Li Meng Yan, la chercheuse de Hong-kong réfugiée aux USA, deux sites de restriction encadrent également très précisément le RBM (216 nt) du SARS-Cov2 (voir chapitre suivant) ce qui pose question.
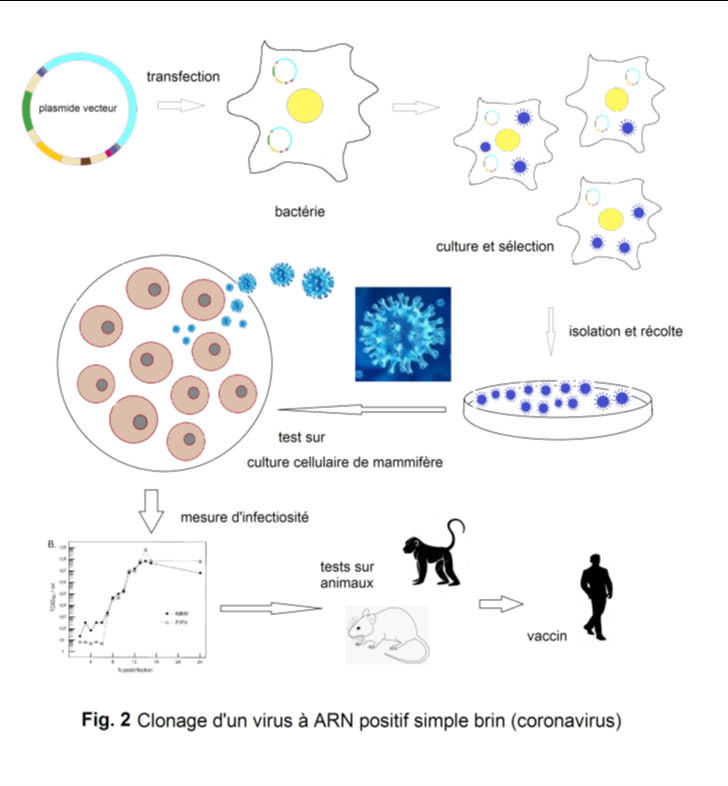
Toutes ces techniques permettent à l’heure actuelle la création et la manipulation complète d’un génome viral dans un intervalle de temps de l’ordre de quelques mois à une année, selon la capacité du laboratoire et le nombre de laborantins y travaillant. Elles ne laissent pas forcément de traces au contraire de ce que des chercheurs de l’Institut Pasteurs prétendent parfois à leurs interlocuteurs candides. Cela a conduit à l’élaboration de recettes bien établies et les techniques fonctionnent parfaitement en routine. La compétence humaine nécessite une bonne formation technique en microbiologie, mais surtout de la minutie et de la patience dans l’application rigoureuse des recettes. Ces qualités indispensables conviennent parfaitement aux jeunes femmes que l’on trouve en très grand nombre comme techniciennes, chercheurs et savantes émérites dans ce domaine de la science.
Il a fallu 5 à 6 décades de recherche et de génie scientifique pour découvrir et comprendre les mécanismes moléculaires essentiels du vivant et leurs applications. De façon générale, cette connaissance et les possibilités qui en découlent (en mal comme en bien), par exemple en termes de dépistage et de vaccination, échappent au grand public qui ne perçoit que confusément ce qui se passe. Le grand danger est de croire que la science du vivant peut se réduire de façon technocratique à un ensemble d’actions et de réactions sur un mode binaire (on-off) entièrement contrôlable. Cet état d’esprit a conduit le gouvernement français, sous influence des puissances financières, à la déraison au cours de l’épidémie. Le degré de complexité est tel que les journalistes sont incapables de mener correctement une interview dans ce domaine. Ils n’ont pas d’autre choix que de suivre la doxa imposée par les chefs de rédactions en symbiose intellectuelle avec les cercles fermés du pouvoir financier.
Partie 2 — Avènement en 2012 de l’ère de l’édition génomique sans restriction
Nous avons vu, dans la partie 1 du chapitre, que des manipulations génétiques de coronavirus ont eu lieu depuis une vingtaine d’années entre autres dans le but de vérifier les facteurs qui pouvaient leur faire franchir la barrière des espèces. Nous nous proposons dans cette partie du chapitre de donner quelques explications, loin d’être exhaustives, sur les techniques utilisées dont le « grand bond en avant » a eu lieu à partir de 2012 avec l’invention de la technique CRISPR-Cas9, dite des ciseaux moléculaires, pour laquelle les professeurs Emmanuelle Charpentier et Jennifer Doudna ont reçu le prix Nobel de Chimie cette année. Mais leur découverte est déjà surpassée par les chercheurs chinois, de l’Université des Sciences et Technologies de la Province de Hebei, qui ont mis au point en 2016 une technique (NgAgo) encore plus performante, inspirée de CRISPR-Cas9.
Comme nous le verrons, il y une ironie déconcertante dans cette coïncidence de l’attribution de ce prix l’année même de l’apparition d’une pandémie dont l’origine naturelle reste mystérieuse, ouvrant les portes à la possibilité d’un accident de laboratoire. Était-ce volontaire de la part du Comité Nobel pour attirer l’attention du Monde sur cette technologie révolutionnaire qui permet assurément toutes les manipulations de virus dans le domaine du gain de fonction ? Les modifications engendrées sur les génomes des organismes vivants manipulés avec cette technique ne laissent aucune trace, ouvrant la porte de façon confidentielle aux déviations les plus fantasques. Les journaux et les grands médias télévisuels français se sont gardés de s’étaler sur une nouvelle qui a déjà changé le monde sans que personne, ou presque, ne s’en aperçoive. À tel point, que le journal Nature qualifie cette découverte de révolution tranquille. Cette découverte nous fait rentrer de gré ou de force dans un monde, qui il y a encore 10 ans, aurait relevé de la science-fiction. Cet avènement augmente considérablement (de façon quasi infinie) le champ des possibilités en thérapie génique pour remplacer un gène défectueux provoquant des maladies héréditaires bien connues, des troubles fonctionnels ou mécaniques.
Évidemment, en premier lieu cette avancée spectaculaire trouve des applications directes au niveau de la manipulation à gain de fonction GOF des virus. Nous soulignons ici l’intérêt immense que la manipulation GOF de virus offre dans la lutte contre le cancer. Certains virus peuvent être en quelque sorte programmés et acquérir ainsi la capacité de détruire spécifiquement des cellules cancéreuses (réf1, réf2, réf3). Par exemple, un variant peu pathogène du virus de la rage s’est révélé particulièrement virulent contre les cellules cancéreuses, mais peu actif dans les cellules normales. Cependant, cette possibilité très prometteuse n’est pas encore transformée en réalité et les recherches sur le gain de fonction curative progressent, semble-t-il, bien plus lentement que celles sur les gains de fonction infectieuse. Mais laissons ici le lecteur à sa réflexion et poursuivons du côté plus obscur.

Avec la découverte de la technique de transgenèse CRISPR-Cas9 l’utilisation d’enzymes de restriction pour couper une séquence d’ADN n’est plus nécessaire. Ces coupures peuvent dorénavant se faire à volonté à un certain nombre d’endroits d’un gène, sans la nécessité de la présence de sites de clivage par enzyme de restriction en amont et en aval du site cible de la manipulation. On dit que cette nouvelle technique ne laisse pas de trace parce qu’elle n’est pas tributaire, comme nous l’avons vu précédemment, de la présence de ces sites de clivage. En réalité, du fait des nombreuses enzymes de restriction connues, il y a un nombre considérable de sites de clivage possibles le long d’une séquence d’ADN et donc on pouvait déjà faire auparavant des mutations qui semblent naturelles. Le problème était plutôt de cliver l’ADN en un endroit unique sans qu’il soit découpé en plusieurs morceaux. Cela restreignait le champ des possibilités ou forçait à faire des opérations multiples pour contourner la difficulté.
En fait, la technique CRISPR-Cas9 peut se rapprocher du système basé sur les enzymes de restriction, mais de façon beaucoup plus sophistiquée. Elle fait appel également à une nucléase bactérienne (procaryote) Cas9 qui participe à un mécanisme de défense très performant des bactéries contre les virus. « Un grand nombre d’entre elles possèdent dans leur génome de courtes séquences d’ADN répétées régulièrement, appelées CRISPR (pour Clustered Regularly Interspaced Short Palindoromic Repeats [NDA, des courtes séquences palindromiques regroupées et espacées régulièrement]). Elles permettent aux bactéries de conserver la mémoire d’une infection par un virus pour mieux s’en défendre les fois suivantes. » À la suite d’une infection, des séquences d’ADN du virus sont intégrées au sein de ces séquences CRISPR. Lors d’une nouvelle infection par le virus, l’ADN viral au sein des CRISPR est recopié en ARN qui se lie à la protéine Cas9 qui ensuite peut reconnaître l’ADN du virus grâce à ce guide et le cliver pour le détruire.
Les travaux d’Emmanuelle Charpentier et Jennifer Doudna à l’Université de Berkeley, publiés en juin 2012, et par la suite de Feng Zhang du Broad Institute (Massachusetts Institute of Technology, MIT) en janvier 2013, ont montré comment cette méthode peut être appliquée à n’importe quelle cellule eucaryote, c’est-à-dire de mammifère.
Ces découvertes ont fait l’objet de dépôts de brevet menant à une âpre dispute entre l’Université de Berkeley et le Broad Institute, car les techniques qui en découlent, à l’instar de la PCR, vont rapporter des centaines de millions de dollars, voire des milliards, aux détenteurs des brevets.
L’importance de cette découverte capitale aux retombées incalculables pour les sociétés humaines et leur environnement a fait l’objet d’une première longue note du ministère des Affaires étrangères le 18 décembre 2015. « Outre la possibilité de modifier le génome de n’importe quelle espèce, plus facilement et à moindre coût qu’avec les techniques précédentes, le système CRISPR/Cas9 (voir plus bas) a permis le développement d’approches inédites en ingénierie génétique dont les retombées sont loin de se limiter à la sphère académique. » Elle pourrait à terme contribuer à éliminer certaines maladies, mais suscite des questions sur leur dérive potentielle vers l’eugénisme. On ne peut s’empêcher ici de remarquer la coïncidence, la même année, entre le prix Nobel décerné et le vote en catimini de la loi sur la bioéthique, dans la nuit du 31 juillet, par 60 députés LERM. Cette loi officialise l’avancée de ces dernières années dans le domaine des manipulations génétiques rendant possible une forme de transhumanisme génétique. Elle donne le feu vert en France à toutes les possibilités de manipulations dans le domaine de la reproduction humaine, aux conséquences incalculables. En 2018, des chercheurs chinois ont annoncé avoir réussi à faire naître des jumelles dont l’ADN avait été modifié pour les protéger du virus VIH dont leur père est porteur. Selon John Christodoulou, de l’Université de Melbourne la technique CRISPR-Cas9 a été utilisée. L’eugénisme génétique est dangereux du fait qu’il va certainement réduire à terme le pool génétique de l’humanité et engendrer une forme de « consanguinité » qui peut mener la dégénérescence ou l’extinction de l’espèce. Le gène de résistance au virus du SIDA a été repéré en Scandinavie et en Russie, conférant protection naturelle contre le virus chez 16 % et 13 % de la population de ces pays, les lymphocytes CD4+ résistant au VIH grâce à l’absence d’un des co-récepteurs à leur surface.
Supposons que l’on confère cette résistance à l’humanité entière, en éteignant comme chez les jumelles chinoises le gène de ce co-récepteur. On va de ce fait réduire son pool génétique, la laissant peut-être sans défense contre un autre agent pathogène dans le futur. À quoi sert naturellement ce co-récepteur ? Il a forcément une fonction potentielle. Rien, n’est plus dangereux que de vouloir se substituer aux lois de l’évolution qui incluent la co-évolution harmonieuse des espèces.
Fait peu connu des pays occidentaux, le Sida est un fléau en Chine dont on a du mal à délimiter les contours tant la maladie est honteuse. En 2010, le nombre de cas était estimé à 12 millions alors que la Chine ne donne plus d’information sur le nombre de cas confirmés par son système de santé depuis 2003. Malgré 700 000 décès du Sida par an, l’épidémie mondiale semble avoir été contenue. Mais à terme, dans quelques décennies, peut-être bien à un horizon plus lointain, le virus du Sida peut poser problème s’il se répand inexorablement, comme cela semble se passer en Chine. Il pourrait réduire dramatiquement la population terrestre et créer une réduction du pool génétique humain à celui de la fraction des populations résistantes. Nous ne pensons pas que ce scénario se passera, car les deux modes de transmission du virus (par le sang et les rapports sexuels) sont facilement contrôlables, en principe. Il serait pourtant tentant pour certains de mettre fin à ce fléau par transgénisme. Cependant, nous mettons en garde ceux qui penseraient que le transgénisme aura la réponse à tous les maux de l’humanité. Certes, il pourra résoudre certains problèmes ponctuels, mais utilisé massivement il en créera d’autres aux conséquences incalculables. Aux USA, une invention a été faite de munir les petits avions de tourisme d’un parachute en cas de panne de moteur, mais cela réduit les crashs que si on continue de maintenir les avions avec la même rigueur. De même, avec les voitures possédant des freins ABS, on ne réduit pas ses risques d’accident si on en profite pour rouler plus vite ou moins précautionneusement. L’emploi sécurisé des techniques d’édition de génome ne peut donc être fait dans un climat de déréglementation sauvage.
Pour clore cette partie, il suffit de rappeler ce que nous mentionnions en exergue et décrit par Yvonne Tran, la rédactrice scientifique du site gouvernemental Diplomatie France.fr, c’est-à-dire que cette avancée a été déjà surpassée par des chercheurs chinois de l’Université des Sciences et Technologies de la Province de Hebei. Ils ont mis au point en 2016 une nouvelle technique (NgAgo) possédant un certain nombre d’avantages comparé à CRISPR-Cas9 dont elle s’inspire. Elle permet par exemple de cibler un plus grand nombre de sites éditables sur l’ADN, pratiquement tous. De plus, les probabilités d’erreur de guidage sont plus faibles, rendant la technique beaucoup plus précise que CRISPR-Cas9 et plus efficace par la même occasion. NgAgo ouvre ainsi un nouveau champ de possibilités pour l’édition génomique et se présente comme un sérieux concurrent de CRISPR-Cas9. » (Yvonne Tran)
Partie 3 — Quel rôle l’Institut Pasteur a-t-il joué dans le contrôle de l’épidémie en France ?
Lorsque les chercheurs de l’Institut Pasteur ont été questionnés sur la possibilité que le SARS-Cov2 soit un virus manipulé, ils ont répondu que si c’était le cas des vestiges de manipulations devraient se retrouver dans le génome. Ils entendaient par-là que, dans le cas d’un accident non intentionnel de laboratoire, la présence de sites de clivage d’enzymes de restriction devrait révéler la manipulation. Cependant, comme nous l’avons montré dans la première partie de ce chapitre, il est tout à fait possible depuis le début des années 2000 de manipuler des coronavirus entiers sans laisser de trace. Ils sont restés très discrets également sur la nouvelle technique de manipulation génétique CRISPR-Cas9. Elle permet ce que l’on appelle à présent l’édition de génome, tant la comparaison avec les logiciels qui permettent la modification rapide et simple d’un texte est tentante. Ils ne pouvaient pas l’ignorer. D’ailleurs, notons qu’Emmanuelle Charpentier est un peu l’enfant prodige de l’Institut Pasteur puisqu’elle y a poursuivi des recherches doctorales entre 1992 et 1995.
Tout un chacun a pu constater le silence assourdissant de l’Institut Pasteur depuis le début de l’épidémie et les quelques réponses éparses obtenues sur la nature et l’origine du virus n’ont été que trop évasives et passablement trompeuses. Par exemple, l’affirmation que le virus SARSCov2 était très distinct du SARS-Cov est de toute évidence une contre-vérité sur le plan de la structure de son génome, de son organisation spatiale et de son mode de fonctionnement.
Il n’est pas concevable qu’étant donné son rôle dans la détection et la prévention des épidémies, l’Institut Pasteur n’ait pas été au courant très tôt, dès les mois de décembre et janvier, du danger de cette épidémie. Pourquoi les gens de Pasteur n’ont-ils pas alerté les autorités françaises ou tenu une conférence de presse publique dès le courant janvier ?
Selon son site internet, l’Institut Pasteur abrite la « Cellule d’Intervention Biologique d’Urgence (CIBU), été créée fin 2002 sous l’impulsion du Directeur Général de la Santé (DGS) et du Directeur Général de l’Institut Pasteur, afin de répondre aux “urgences biologiques spécialisées”. Ces urgences peuvent être des épidémies, des accidents ou une utilisation potentielle d’armes d’origine biologique, toutes mettant en péril la santé publique. » Les missions de la CIBU incluent :
– mobiliser à l’international de l’expertise en cas d’urgence représentant une menace pour la sécurité sanitaire en France ;
– informer sans délai l’InVS en cas de détection de tout phénomène inhabituel susceptible d’avoir un impact sur la santé publique.
Que faisait son directeur, Jean-Claude Manuguerra, au cours du mois de janvier 2020, quand il a été mis au courant de la situation à Wuhan ? On l’a malheureusement vu déclarer le 21 janvier 2020 sur Euronews à propos de l’épidémie en Chine : « … c’est quelque chose de nouveau, mais qui pour l’instant n’est pas plus préoccupante que ça. Il faut juste ne pas louper le coche pour endiguer ce phénomène épidémique… ». S’il y avait une personne en France qui connaissait le danger de laisser pénétrer en France le SARS-Cov2 c’était bien lui. La séquence du premier isolat WH-Human 1 du coronavirus (appelée aussi 2019-nCoV) et son génome complet (29,903 nt) avaient été déposés le 14 janvier 2020 dans GenBank, sous code d’accession MN908947 avec la définition « Génome complet de l’isolat du virus WH-Human 1 de pneumonie du marché aux fruits de mer de Wuhan ». Donc, la séquence du virus avait forcément été analysée par les équipes de l’Institut Pasteur. Elles savaient à quel point le motif de liaison (RBM) de la protéine S était proche de celui du SARS-Cov de 2002-2003 ainsi que sa polymérase RdRp (96,5 % d’identité) signant la dangerosité du virus. De plus, les autorités chinoises annonçaient le caractère contagieux du virus le 20 janvier, avec 1 mois de retard, alors que les autorités de Wuhan connaissaient le caractère contagieux depuis la mi-décembre (chapitre 1). Le 24 janvier, il était publié dans le Lancet que le virus était transmissible de personne à personne. Le directeur de la CIBU savait donc le 21 janvier, lors de son interview sur Euronews, que ce nouveau virus était de type SARS-Cov avec tout ce que cela implique. Qu’a-t-il fait pour informer et alerter les pouvoirs publics avec conviction durant la dernière semaine de janvier et tout le mois de février ?
Dans la section qui suit, nous découvrons qu’en compagnie de 15 autres chercheurs à l’Institut Pasteur, il est co-inventeur d’un brevet sur les vaccins contre les virus du type SARS-Cov. Ce brevet prouve que Pasteur s’était préparé depuis 2003 à l’éventualité de la ré-émergence d’un virus similaire au SARS-Cov. Ils ont procédé à des milliers d’expériences pour comprendre comment faire des tests de dépistage et des vaccins contre ce type de virus émergeant. Ils ont breveté le résultat de toutes ces expériences pour être en mesure, le cas échéant, de tirer les bénéfices financiers potentiels d’une épidémie de SARS-Cov qui ne serait pas maîtrisée à temps, nécessitant dépistage et vaccination massive. Cela, les dispensait-il de leur rôle d’alerte des pouvoirs publics ?
Les brevets de portée mondiale de l’Institut Pasteur
L’Institut Pasteur détient depuis 2005 des brevets de portée mondiale (EP 1 694 829 B1 et US 2007/0128224 A1) sur les coronavirus à SARS. Ces brevets qui courent au moins jusqu’en 2027 ont pour but d’explorer et de décrire le maximum de caractéristiques de façon à établir des vaccins ou des tests de dépistage sur des coronavirus de type SARS-Cov. Ces brevets se basent sur le séquençage d’un isolat complet du virus du SARS-Cov récupéré à la Clinique Française de Hanoï en février 2003. Cela était parfaitement légitime pour étudier la confection de vaccin contre l’éventualité d’une épidémie de coronavirus à SARS du type de celle de 2002-2003 dans les années à venir. Cependant, soyons clairs, le seul but d’un brevet est l’argent qui est le facteur numéro 1 de survie pour les instituts de recherche. Mais cela amène une grande confusion, et permet d’introduire le biais du conflit d’intérêts dans un domaine particulier de la santé publique (les épidémies) qui devrait en être intégralement exempt. En effet, les intérêts individuels sont indissociables des dépôts de brevet en termes de promotion de carrière et d’avantages financiers pour les inventeurs et leurs laboratoires de recherche.
À ce sujet notons d’ailleurs que dans un premier tant l’Institut Pasteur s’était fait doubler par le Center for Disease Control (CDC) d’Atlanta qui avait récupéré un premier isolat à la Clinique Française de Hanoï qui a conduit à un brevet séparé, US 7220 852 B1, déposé par des chercheurs américains pour l’élaboration de tests de dépistages. Mais le brevet de Pasteur va bien plus loin puisque son but était de breveter en plus de tests de dépistage, par PCR et par anticorps monoclonaux, des méthodes de vaccination.
On raconte toujours au grand public qu’il n’est pas possible de déposer de brevet sur ce qui a trait au vivant, comme des virus ou des protéines d’origine naturelle. En réalité cela n’est pas exact, il est tout à fait possible de le faire à condition que le brevet s’accompagne de descriptions des méthodes inventées pour manipuler l’élément moléculaire appartenant au vivant. Tout entreprise ou organisme se situant dans la zone géographique couverte par le brevet, et utilisant une ou des méthodes décrites par le brevet, se verront dans l’obligation de payer des royalties au titulaire du brevet. Dans le cas de tests de dépistage et de vaccins généralisés sur une zone couvrant l’UE et les USA les retombés se comptent en centaines de millions d’euros, au minimum (voire des milliards sur la population des 27 pays de l’UE qui comptent 400 millions d’habitants). Il convient de préciser également que, contrairement à ce qui est dit souvent, les brevets US de portée mondiale sont purement nominatifs, c’est-à-dire que l’invention est attribuée obligatoirement à une ou des personnes physiques (les chercheurs qui ont inventé les méthodes brevetées) et non à des personnes morales (comme des sociétés ou instituts de recherche). En ce qui concerne les brevets européens, des personnes morales peuvent être titulaires du brevet sans toutefois abroger les droits des chercheurs inventeurs, inaliénables au regard du droit international. Par exemple, l’Institut Pasteur, le CNRS et l’Université Paris VII sont les titulaires du brevet EP 1 694 829 B1, mais une liste de 16 inventeurs nominatifs est précisée ensuite.
Nous ne nous étendrons pas sur les noms, mais nous avons parfaitement le droit de les citer. Ces documents ne sont absolument pas confidentiels et, en droit, doivent pouvoir être consultés par quiconque. Par contre, il est évident que toute publicité autour de ces brevets gêne terriblement l’Institut Pasteur. Pourquoi ? La raison en est, comme nous l’avons dit, que les droits des inventeurs ne peuvent pas être totalement abrogés par les titulaires officiels du brevet. En d’autres termes, des clauses contractuelles entre l’Institut Pasteur et les inventeurs du brevet peuvent très bien exister, probablement comme il se pratique couramment aux USA. N’oublions pas que l’Institut Pasteur déclare être une fondation privée à but non lucratif consacrée à l’étude de la biologie, des micro-organismes, des maladies et des vaccins. Comment, son conseil d’administration répartit-il les revenus des brevets dont ses chercheurs sont les inventeurs ?

Nous posons la question au ministre de la Santé : Madame Sylvie Van der Werf, directrice d’un département de recherche à l’Institut Pasteur et premier inventeur sur ces brevets, qui siège au HCSP, peut-elle donner un avis en toute indépendance sur les mesures concernant la gestion de l’épidémie en France ? Les décisions prises par les autorités françaises peuvent avoir un effet d’entraînement sur les 26 autres pays de l’UE et les USA et rapporter des sommes colossales. Notons que 15 autres chercheurs de Pasteur sont également co-inventeurs des patentes en vigueur, dont un intervient sur le plateau de BFMTV. Les brevets européen et américain de Pasteur couvrent les tests de dépistages par PCR et par anticorps monoclonaux du SARS-Cov2. Couvrent-ils également les vaccins mis au point si rapidement ? Ils décrivent un éventail très large de méthodes spécifiques d’élaboration de vaccins contre les coronavirus de type SARS. Dans quelle mesure ces brevets couvrent-ils les vaccins classiques que l’on va proposer aux Français, en dehors de la thérapie génique par ARN messager. Cela constitue-t-il un conflit d’intérêts quand la décision est prise de faire plus de 1 million de tests de dépistage par semaine sur plusieurs mois, ou si un jour il est décidé de vacciner massivement le peuple français ? Les Français ont le droit de savoir.
Beaucoup s’étonnent de la rapidité avec laquelle des vaccins ont pu être mis au point, car en principe cela prend plusieurs années. Mais c’est de la naïveté de croire que certains groupes industriels et de recherche ne s’y étaient pas préparés à l’avance, à l’instar de l’Institut Pasteur. Depuis plusieurs années, certains milieux avaient anticipé ce qui allait se passer inéluctablement, car après 2003 aucune mesure sérieuse n’avait été prise par la Chine pour fermer ou limiter l’activité des marchés aux animaux sauvages. Les vaccins classiques qui vont être proposés (pas ceux à ARNm) ou imposés ont pu être préparés en quelques mois grâce aux recherches reliées au brevet de l’Institut Pasteur.
Ce que contiennent les brevets de l’Institut Pasteur
Pour le lecteur candide, nous devons expliquer que le but d’un brevet est de bloquer un certain nombre, le maximum en fait, d’utilisations possibles autour une application ou une invention décrite. Par exemple, en chimie on ne patente pas une molécule, mais toutes les voies de synthèse chimique permettant de la produire. Évidemment, si quelqu’un découvre une nouvelle voie de synthèse il a le droit de déposer un brevet également sur cette molécule. L’analyse du brevet de l’Institut Pasteur montre comment ce principe est utilisé pour essayer de garantir qu’il couvre le plus possible, en matière de tests dépistage et de vaccination, les autres virus de type SARS-Cov. Les pseudo-vaccins à ARN messager qui vont être utilisés ne tombent pas sous le coup de ce brevet. Ils font appel à un principe de thérapie génique jamais expérimenté auparavant sur l’être humain.
Les brevets US 2007/0128224 A1 et EP 1 694 829 B1 de l’Institut Pasteur sont strictement équivalents, mais recouvrent 2 zones géographiques distinctes. On y trouve en première (brevet US) et dernière partie (brevet EU) la description détaillée du séquençage et de la caractérisation du SARS-Cov à partir de l’échantillon d’Hanoï. On trouve aussi un arbre phylogénique qui établit que le virus n’est pas le résultat de mutations ou de recombinaisons d’un virus proche et appartient donc à une classe nouvelle SARS-Cov, appelée du nom du syndrome qu’il a engendré. Cet arbre a son importance pour la couverture des applications du brevet. Cela signifie : « ce que nous décrivons dans ce brevet s’appliquera à tout virus de type SARS-Cov ». On trouve également sur 80 pages la description détaillée de la séquence du virus, du nucléotide 1 à 29746, qui cartographie l’intégralité des sites de clivage par enzymes de restrictions. Il y en a environ 4000 le long de la séquence.
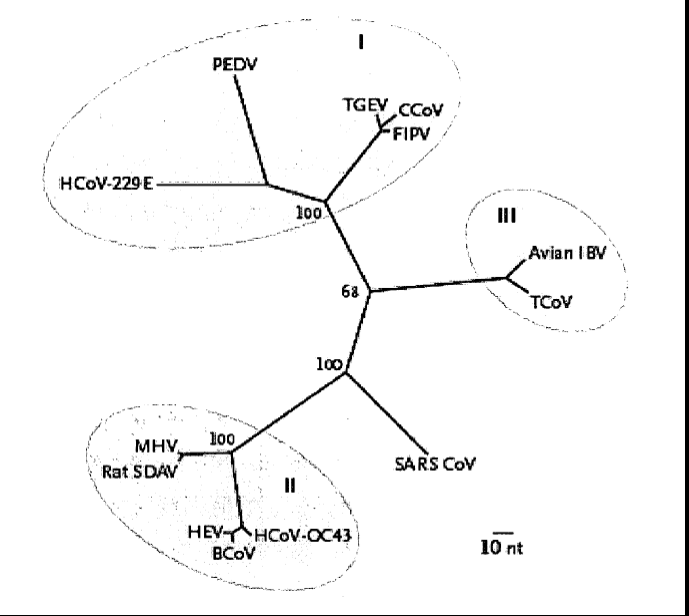
Ils font partie de la description des applications brevetées. La présence de ces sites en très grands nombres est naturelle. Elle est due au fait qu’il y a des centaines d’enzymes de restriction connues ce qui permettrait finalement de pouvoir effectuer des manipulations à n’importe quel endroit du virus. L’objet du brevet est que ces milliers de sites de clivage identifiés permettent de découper le virus en fragments qui peuvent servir d’amorce pour les tests PCR, soit pris séparément soit par groupe de plusieurs. Au cas ou certains douteraient que cette partie du brevet puisse s’appliquer aux tests de dépistage développés pour le SARS-Cov2, nous les rassurons, cela peut s’appliquer dans nombre de cas par ce que le point de blocage [0051] du brevet le stipule : « Lesdits fragments sont, soit sous forme de fragments isolés, soit sous forme de mélanges de fragments. Les Inventeurs décrivent également des fragments modifiés, par rapport aux précédents, par enlèvement, ou addition de nucléotides dans une proportion d’environ 15 %, par rapport à la longueur des fragments ci-dessus et/ou modifiés au niveau de la nature des nucléotides, dès lors que les fragments nucléotidiques modifiés conservent une capacité d’hybridation avec les séquences d’ARN génomiques ou antigénomiques de l’isolat tel que défini ci-dessus. »
Le brevet couvre donc absolument tous les tests PCR qui pourraient être faits sur un coronavirus qui aurait jusqu’à 15 % de différence d’identité de séquence avec le SARS-Cov. Au niveau du génome complet le virus SARS-Cov2 n’est qu’à 80 % identique au SARS-Cov, cependant les parties d’intérêt de la séquence, spécifique à l’identification d’un virus de type SARS, comme en particulier la polymérase RdRp est à 89 % identique à celle du SARS-Cov et donc les amorces PCR des tests pour sa détection sont très certainement couvertes par le brevet. De même les protéines E, M et N de la capside, de la membrane et de l’enveloppe du SARS-Cov2, avec 95 %, 85 % et 89 % d’identité sont couvertes par le brevet.
Les brevets européen et américain contiennent respectivement 400 et 600 points de blocages identifiés au cours des milliers d’expériences réalisées par l’Institut Pasteur. Les blocages concernent : (a) les tests de dépistage par PCR ; (b) des tests de dépistage par anticorps monoclonaux ; (c) des techniques de clonage et d’expression de l’intégralité ou de parties du virus dans l’optique d’élaboration de vaccins contre les virus de type SARS-Cov. Ce troisième aspect du brevet, le plus considérable, est extrêmement complexe à analyser pour un non spécialiste. Nous invitons les experts virologues désireux d’expliquer en quoi les brevets peuvent couvrir les vaccins fabriqués ou en cours d’élaboration à communiquer avec France Soir.
En résumé, l’Institut Pasteur a dépensé une énergie considérable en ressources humaines, en argent et en matériel pour faire des études dans le but de démonter et bloquer l’intégralité des voies de vaccination possibles contre ce type de virus. Le dépôt et la mise à jour des brevets ont nécessité des années d’effort et ont coûté des millions d’euros, juste en ce qui concerne les frais reliés à l’établissement des brevets, sans parler des frais de recherches techniques. En raison des nombreux collaborations et liens entre les chercheurs de l’Institut Pasteur, de Sanofi-Mérieux et ceux de l’Institut de Virologie de Wuhan, l’Institut Pasteur ne pouvait ignorer dès la fin décembre 2019 ce qui se passait à Wuhan et la catastrophe planétaire que cela pouvait engendrer.

On ne dépense pas autant d’effort et d’argent pour déposer des brevet d’une durée de validité de 20-25 ans sans avoir dans la tête qu’une épidémie risque très probablement de surgir dans cet intervalle de temps.
Conclusion
Un test de dépistage par PCR coûte 54 euros à la Sécurité Sociale, donc au contribuable. À raison de 2 millions de tests de dépistage effectués par semaine à partir de début novembre (et aux alentours d’un million courant octobre) cela représente un chiffre d’affaires de 108 millions d’euros/semaine pour les fabricants de tests. Les tests sont d’une utilité assez illusoire puisque de toute façon une fois déclaré positif on ne reçoit aucun traitement mis à part rester chez soi pendant 2 semaines, en se mettant en arrêt maladie, et attendre une éventuelle aggravation. L’immense majorité des personnes testées n’ont pas de symptôme, ou très peu. Personne ne le dit, mais ces tests ont conduit à une très grande quantité d’arrêts de travail, alors que la positivité au test était dans de trop nombreux cas, soit fausse, soit le résultat d’une infection non-symptomatique remontant à plusieurs semaines. En cas d’arrêt, l’employeur et la Caisse Primaire d’Assurance Maladie (CPAM) sont mis à contribution lourdement. Ne parlons même pas du fait qu’au-delà 30 cycles d’amplification la fiabilité des tests diminue de façon outrageuse (ref1, ref2) .Un tel système n’a fait que conduire les Français, de façon plus ou moins consentante, a une paranoïa déraisonnable et la ruine programmée de leur pays. Quelles sont les sociétés pharmaceutiques ou les biotechs qui produisent ces tests en France ? Quelles sont leurs relations avec l’Institut Pasteur ? Combien cela rapporte-t-il à l’Institut Pasteur ? Le brevet de l’Institut Pasteur couvre-t-il les vaccins qui vont être proposés ou imposés aux Français dès la fin du mois de décembre ou le mois prochain ? Autant de questions auxquelles il serait opportun de pouvoir répondre en sachant qu’une directrice de l’Institut siège au HCSP.
Chapitre 8 — Et si le SARS-Cov2 était le résultat raté d’un vaccin conçu contre le VIH et le paludisme ?
Comme nous l’avons vu au chapitre précédent, depuis le début des années 2000 on sait manipuler un génome complet de coronavirus, en particulier on sait échanger et manipuler le domaine S1 de la protéine de pénétration cellulaire qui est au cœur du débat. Il est à noter que tous les articles universitaires décrivant les découvertes et les mises au point des techniques publiées depuis 20 ans dans ce domaine impliquent des chercheurs d’origine chinoise. Ils se retrouvent toujours en grand nombre dans la liste des auteurs et quasi systématiquement au premier plan comme premiers auteurs. Les manipulations de microbiologie et le temps nécessaire aux recherches dans ce domaine impliquent un nombre considérable d’heures passées dans les laboratoires. En fait, il s’agit d’y passer le plus clair de son existence, souvent au-delà de 10 heures par jour, 7 jours sur 7. Depuis 30 ans, les étudiants chinois ont eu la patience et le courage de se plier à cet esclavage dans les pays occidentaux, en particulier les USA, pour parvenir à un niveau de compétence qui leur assure une avancée sociale. Cela leur a également permis de ramener en Chine la maîtrise des techniques de manipulation les plus performantes. Soyons clair, la Chine n’a pas eu plus de complexes dans l’exploitation de ces techniques que les pays occidentaux n’ont en eu dans l’exploitation des étudiants chinois. Il est incontestable que la Chine s’est livrée depuis une décennie au moins à de multiples recherches sur l’élaboration de virus vaccinaux de synthèse contre le SARS, mais aussi très certainement contre d’autres maladies qui sont des fléaux comme le sida et le paludisme.
Partie 1 — Les vaccins à ARN messager et le lobby pharmaceutique risquent de nous faire passer de Charybde en Scylla
Le fléau du lobbying pharmaceutique a pesé sur le non-traitement de l’épidémie en France. À présent, le fléau désigné par les autorités est le SARS-Cov2 et la folie vaccinale a saisi les gouvernants occidentaux (UK, EU et USA), faisant fi des possibilités existantes par traitements antiviraux à usage prophylactique chez les personnels à risque ou dès les premiers symptômes chez l’ensemble de la population. Il serait presque inutile de rappeler ici le bénéfice avéré des traitements à base d’HCQ et d’azithromycine tant la population française est à présent consciente d’avoir été manipulée. Nous remercions le combat du Pr Raoult à l’IHU Méditerrané et celui de bien d’autres praticiens connus comme le Pr Perronne, ou bien médecins anonymes, pour faire connaître dans notre pays le mérite de ces traitements très peu coûteux (moins de 10 euros par patient). Ces deux professeurs font face à des ennuis qui relèvent d’un régime de dictature. France Soir a fait paraître depuis plus de 6 mois d’innombrables articles (ref1, ref2, ref3, ref4, ref5…) relatant ce combat pour la vérité contre les puissances de l’argent qui dominent la France.
Ces molécules ne sont pas les seules à pouvoir améliorer grandement le tableau clinique de cette épidémie. Citons également l’ivermectine un médicament antiparasitaire surpuissant, contre la gale et les parasitoses intestinales, dont les propriétés antivirales sont en train d’être découvertes ou redécouvertes depuis 6 mois. Il est très efficace sur les parasitoses, même aux stades les plus avancés, et il fait baisser radicalement les niveaux de SARS-Cov2 en 48 heures. Une prise orale unique ajustée au poids de la personne suffit (coût du traitement < 10 euros). Selon les études cliniques randomisées effectuées depuis 6 mois et publiées, ce médicament réduit de 3 à 4 fois la mortalité hospitalière reliée au SARS-Cov2. Pourtant, les autorités de santé sollicitées refusent d’autoriser sa prescription dans le cadre de la lutte contre l’épidémie, que ce soit en prophylaxie, en traitement précoce ambulatoire, ou en traitement hospitalier. Nous supposons que le prétexte est que cette molécule peut comme tout médicament actif engendrer des effets secondaires potentiellement toxiques, mais très rares qui concernent les patients parasités. Il s’agit en l’occurrence principalement d’atteintes hépatiques dont la sévérité est variable et le plus souvent reliée à la densité parasitaire. Les effets indésirables sont dans la plupart des cas légers et transitoires, mais leur incidence et gravité peuvent être majorées chez les sujets poly-parasités. Selon le Vidal, un effet très dangereux, mais très rare (< 0.01 %) est la nécrolyse épidermique toxique, qui peut être observée aussi avec de très nombreux médicaments surdosés. Bref, cela montre que ce médicament très puissant ne peut pas être prescrit en prophylaxie généralisée, mais pourrait parfaitement être utilisé de façon encadrée au moins en milieu hospitalier.
J’ai travaillé pendant 13 ans à l’élaboration d’essais cliniques dans le domaine de la cancérologie et j’ai pu observer que des effets secondaires médicamenteux extrêmement délétères pour le patient étaient systématiquement minimisés, voire passés sous silence, tant par les investigateurs cliniques que par les autorités de santé, mettant en avant systématiquement les bénéfices pour le patient. Certains traitements d’une toxicité effrayante sont autorisés au prétexte qu’ils rallongent l’espérance de vie moyenne de deux mois, faisant fi de la qualité de vie du patient. Ce genre de traitements coûte des dizaines de milliers d’euros par patient au système de santé et en rapporte autant à Big Pharma.
Avec le Covid-19, nous assistons à l’inversion totale du paradigme. L’objectif noblement affiché par nos gouvernants est à présent le risque médicamenteux zéro. On pourrait penser que cela relève d’une méconnaissance médicale naïve, car aucun médicament actif n’est dénué d’effets secondaires. Cependant, nous notons que pour le remdésivir ce principe de précaution n’a pas été mis en avant, pas davantage que pour la vaccination par ARN messager de Pfizer et Moderna. Le remdésivir est un médicament dont le développement n’a coûté à Gilead que 150 millions d’euros, et qui a bénéficié d’une campagne éhontée de promotion et de lavage de cerveau rarement égalée par le passé. Son usage, d’un coût de 2000 euros par patient, a été finalement abandonné devant sa difficulté d’administration, 10 perfusions intraveineuses ce qui en soi présente déjà un danger, sa génotoxicité probable et sa très forte toxicité rénale qui était connue depuis le début. L’UE en a quand même acheté pour 1 milliard d’euros à son fabricant sur les deniers des contribuables des pays membres et contributeurs nets. Nous voyons donc que les actions du gouvernement sont dictées par les intérêts pharmaceutiques. Pour promouvoir la vaccination généralisée, celui-ci continue de refuser l’usage des antiviraux comme l’HCQ et l’azithromycine, en préventif et comme traitement précoce, et l’ivermectine en traitement encadré. L’UE a passé commande de 1,9 milliard de doses de vaccins (dont 160 millions chez Moderna pour 5,6 milliards d’euros) auprès de 6 compagnies pharmaceutiques pour les 448 millions d’habitants de l’UE (hors UK), soit 4 doses par habitant, pour un coût exorbitant total de 21 milliards d’euros. Il faut noter d’ailleurs qu’en ce qui concerne ces traitements, le problème politique est le même aux USA, pays des lobbys institutionnels. C’est un secret de polichinelle que les lobbys gangrènent à présent les allées du pouvoir en France.
Le vaccin à ARN messager en plus d’une efficacité incertaine ouvre une boîte de Pandore
Ce vaccin qui injecte l’ARN messager (ARNm) de la protéine de pointe S (spicule) du virus SARS-Cov2 laisse pantois. Les coronavirus de chauves-souris sont très éloignés des humains sur le plan évolutif en raison de notre séparation physique avec une espèce restée confinée dans les lieux les plus reculés. Il n’y a quasiment pas eu de coévolution avec l’homme ces derniers millénaires, et le système immunitaire humain manque d’adaptation pour réagir de façon optimale sur l’ensemble de la population. L’introduction forcée, en deux décennies, des coronavirus de chauves-souris dans l’espèce humaine est certainement le résultat de la folie des 10 000 marchés aux animaux sauvages chinois ainsi que des 20 000 fermes d’élevage de gibier et des trafics de chauves-souris associés. Dans une mesure, que l’on ne connaît pas encore, les laboratoires de recherche conjoints en Chine et ailleurs ont peut-être également contribué à créer une situation potentiellement plus explosive dans le futur que maintenant.
Contrairement à la paranoïa gouvernementale, dont les fondements sont à chercher ailleurs, cette épidémie n’est pas de nature à créer une mortalité excessive. Nous étions à 0.046 % à la fin du printemps. À présent, avec la vague de novembre nous atteignons malheureusement 0.09 % étalé sur une année, touchant essentiellement une population âgée ou très âgée. Rappelons que 1 % de la population française meure chaque année de maladie, d’accidents divers et variés et de vieillesse. L’épidémie perdure en France à cause de l’apparition et la réintroduction régulière de nouveaux variants. Ce prolongement est favorisé par les confinements et l’absence de contrôle des frontières. Au printemps, cela a freiné l’établissement d’une immunité collective, même partielle. Le maintien de l’ouverture des frontières pendant l’été a permis la réintroduction du virus sous forme de variants. Le confinement de novembre démontre que cette approche ne marche pas, ou que très médiocrement, car il est impossible de s’opposer à la diffusion d’un virus une fois introduit sur un territoire. Les confinements sont délétères, ils donnent au virus du temps pour s’adapter et évoluer vers d’autres variants dangereux, en optimisant leur pouvoir de contamination. Il n’est pas interdit de penser que l’absence de confinement et le maintien des frontières fermées dès la fin du mois de janvier ou en février eussent été plus efficaces.
Le fait que l’épidémie perdure va pousser le gouvernement à tenter de forcer la vaccination généralisée par tous les moyens de persuasion. Que se passe-t-il dans la tête du président et de ses ministres pour laisser penser qu’ils croient les vaccins plus exempts de problèmes que les traitements antiviraux.
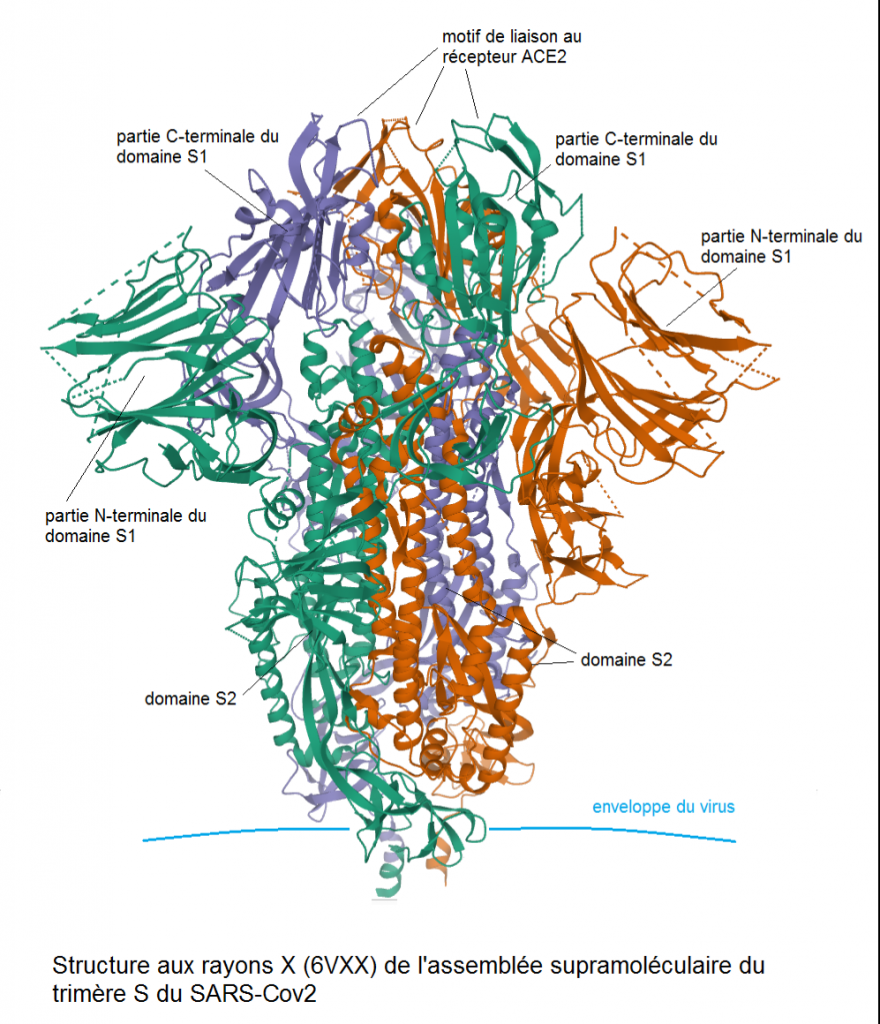
Les effets secondaires, très peu fréquents, des traitements antiviraux sont immédiats alors que la plupart de ceux engendrés par les vaccins vont être largement différés sur des mois et des années sans que l’on connaisse a priori leur fréquence. Il faut également appliquer le raisonnement du ratio bénéfice/risque pour juger du bien-fondé d’un vaccin pour une population donnée. Sur le plan de l’éthique médicale, on ne vaccine que lorsqu’il n’y a pas de traitement alternatif et que le ratio bénéfice/risque escompté l’autorise. Nous voyons que l’éviction des traitements antiviraux permet au gouvernement de privilégier la vaccination généralisée de Big Pharma sans même devoir estimer le ratio bénéfice/risque. En faisant cela le gouvernement choisit qu’il le veuille ou non l’option « après moi le déluge ».
Au-delà des réactions hypersensibles immédiates, qui n’entraînent que très rarement le décès (<1 cas par million), tous les vaccins classiques recèlent des pièges qui ne se dévoilent que plus tard. En particulier, ils déclenchent un certain nombre de réactions auto-immunes à quelques semaines, quelques mois ou quelques années de leur administration. Ce fut le cas avec le vaccin contre le virus H1N1, responsables de cas de narcolepsie en 2009.
En 2010, pas moins de 23,5 millions d’Américains présentaient un diagnostic de maladies auto-immunes selon l’Office of Women’s Health du Ministère de la santé américain. Un chiffre étonnamment élevé qui révèle l’émergence d’un spectre de conditions auto-immunes répandues à l’échelle d’une population entière. Sans pour autant être la cause de l’intégralité de ces troubles, les adjuvants des vaccins sont de plus en plus suspectés d’en être responsables pour une partie non négligeable : troubles auto-immunes rassemblés sous la dénomination ASIA par le National Institute of Health (NIH) aux USA et pouvant apparaître entre 3 jours et 5 ans après l’injection d’un vaccin à adjuvant aluminique.
En ce qui concerne les vaccins à ARN messager, la boîte de pandore est ouverte, car ils relèvent autant du domaine de la thérapie génique que de la vaccination, et le danger vient plus de l’homme et son aveuglement que de la nature.
Le principe de ce vaccin est d’injecter de façon intramusculaire l’ARNm codant la protéine S (spicule) complète responsable de la pénétration dans les cellules pulmonaires par l’intermédiaire de sa liaison au récepteur ACE2. Le gène de la protéine S est la fraction de matériel génétique la plus cruciale du virus puisque c’est celle qui le rend si pathogène en lui conférant un pouvoir de pénétration tout à fait unique dans la lignée des coronavirus à SARS. Les ribosomes des cellules vont transcrire ce matériel génique en protéines S que le système immunitaire va reconnaître comme antigène. Subséquemment, des anticorps spécifiques vont être générés, entraînant également un mécanisme de mémorisation de cette pseudo-infection virale par le système immunitaire. Si par la suite la personne vaccinée est en contact avec le virus, son système immunitaire devrait alors réagir en produisant en grande quantité anticorps dont il aura gardé la mémoire. Nous avons donc bien là en théorie un procédé vaccinal tentant. Cette méthode n’a jamais été testée auparavant sur l’être humain et on ne connaît pas la réalité ni la durée de la réponse immunitaire au-delà des quelques mois de recul des essais conduits récemment.
Cependant, la principale inquiétude avec ce vaccin est qu’il s’agit d’injection de matériel génétique qui peut se retrouver intégré à l’ADN de nos cellules par l’effet de transcriptases inverses. Ces transcriptases peuvent être déjà présentes dans notre organisme à la suite d’une infection par un rétrovirus. Elle peut être asymptomatique ou avoir des effets très différés comme dans le cas du cancer du col de l’utérus (sarcome de Rous) généré par le papilloma virus qui est assez répandu. De plus : « le génome humain comprend naturellement des séquences d’ADN d’origine rétrovirale (8 %). Elles sont les vestiges de l’infection de cellules de nos ancêtres primates il y a plusieurs millions d’années et la plupart de ces séquences semblent inactives. Cependant certaines d’entre elles codent encore des protéines » (Source Institut Pasteur). Cela devrait faire réfléchir nos dirigeants de savoir que des protéines virales introduites dans le génome humain il y a des centaines de milliers, voire des millions d’années, puissent encore être actives.
Des preuves expérimentales que le virus SARS-Cov2 peut être intégré au génome des cellules infectées surexprimant une transcriptase inverse sont pré-publiées sur le site bioRxiv. Cela expliquerait pourquoi certaines personnes restent positives au test PCR du SARS-Cov2 pendant des mois après leur infection. Que va-t-il se passer chez la fraction de personnes vaccinées qui aura intégré la protéine S du SARS-Cov2 dans son génome ? On peut imaginer que l’ARNm de ce gène, sous certaines conditions, va pouvoir être exprimé dans les cellules et pourra, non pas générer le SARS-Cov2, mais créer des réactions d’hypersensiblité auto-immune en déclenchant indûment le système immunitaire. On peut imaginer également que l’ARMm de cette protéine se recombine avec le coronavirus d’un rhume bénin pour créer un nouveau virus du type SARS-Cov… Il ne faut toutefois pas trop s’affoler à l’avance, car, comme nous l’avons vu au paragraphe précédent, notre génome porte déjà de nombreuses traces de virus que nous avons intégrés au cours de centaines de milliers d’années. Cela est plutôt rassurant même si on ne sait pas quel en a été le coût en termes de maladies transmises et auto-transmises et de sélection de populations humaines ou pré-humaines.
En tout cas nous voyons que ce vaccin, au lieu de nous libérer, pourrait risquer de nous rendre dépendants de gestes barrières à vie pour nous et notre descendance ; entre ceux qui seraient devenus un réservoir naturel de coronavirus de type SARS-Cov et les autres pour ne pas propager à leur insu ces nouveaux virus. Bien sûr, il ne faut pas être paranoïaque, loin de là, mais ce genre d’hypothèse doit être formulée en conscience par ceux qui conseillent nos gouvernants. Il est à noter que dans une interview récente le Pr Montagnier nous met en garde contre les vaccins à ARNm, stipulant qu’ils pourraient s’intégrer au génome en restant actifs sur plusieurs générations et, entre autres problèmes, engendrer des réactions auto-immunes ou créer également des cancers. En ce qui concerne les vaccins plus classiques contre le SARS, on sait déjà depuis 2012 qu’ils peuvent engendrer des réactions auto-immunes après immunisation au niveau des poumons lorsque la personne vaccinée est exposée ensuite au virus réel. D’autre part, des réactions adverses sont de toute façon reliées à tout vaccin avec des fréquences d’apparition variables selon les cas. Aux USA, un hôpital de banlieue près de Chicago suspend déjà la vaccination Covid-19 après que 4 personnes sur 3 000 vaccinées ont développé une réaction adverse (tremblement et élévation du rythme cardiaque).
Cerise sur le gâteau, qui démontre le côté potentiellement illusoire de cette vaccination généralisée qui se profile, est que l’on arrive à une phase où le virus mute beaucoup, en particulier dans la région du domaine S1, qui pourrait rendre rapidement inopérant les vaccins. La direction de Moderna ainsi que le gouvernement indiquent déjà que les gestes barrières (masques et distanciation) resteront obligatoires même après la vaccination ! C’est dire le degré de confiance que ces gens ont dans leur vaccin. La Grande-Bretagne semble avoir accéléré, dans une fuite en avant, la vaccination, car un mutant se répandrait dans le sud-est de l’Angleterre plus rapidement que les souches précédentes. Il est aussi étrange que l’UE précipite et accélère sa campagne Pfitzer. La perte d’efficacité vaccinale est une possibilité à prendre en compte, mais en réalité l’immunité croisée entre les différends variants existe en général dans une très large proportion. Donc, un vaccin efficace devrait le rester pour un temps suffisant sur une période de 6 mois à un 1 an. Ainsi, le Pr Raoult observe que seulement 18 patients sur les 10 000 traités à l’IHU auraient développé une seconde fois le Covid-19 au contact des nouveaux variants introduits pendant l’été. Cela est très rassurant et indique que cahin-caha on se dirige vers l’immunité collective et la fin de l’épidémie, indépendamment d’une vaccination généralisée. De toute façon, les vaccins à ARNm ne sont absolument pas indispensables et leur application relève de l’expérimentation humaine à grande échelle.

Pour conclure cette partie et introduire les suivantes, qui traitent des arguments scientifiques développés autour de l’origine possible du virus, nous soulignons des faits remarquables concernant les mutations qui apparaissent. Elles se concentrent en 3 endroits particuliers du génome du virus codant la protéine S, dont 2 concernent le domaine S1.
Une large section du domaine S1, incluant toute la partie N-terminale du domaine S1 et le domaine de liaison au récepteur ACE2 jusqu’au motif de liaison, avait été repérée dès le mois de janvier, par le chercheur universitaire James Lyons Weiler, comme une région à part dans le génome du SARS-Cov2. En effet, elle représente seulement 1,4 % de celui-ci, mais est très divergente sur le plan évolutif par rapport au coronavirus le plus proche qui était à ce moment-là le virus Bat-Cov-ZC45 identifié par la recherche militaire chinoise. De cette observation, Lyons Weiler en a déduit l’hypothèse, sévèrement réfutée par l’Institut Pasteur de Shanghaï, que cette région pouvait avoir été manipulée génétiquement. Il n’était pas interdit de le penser étant donné que le domaine S1 des coronavirus a été l’objet de manipulations constantes depuis une vingtaine d’années. Nous étudierons cette hypothèse dans la partie suivante.
Le troisième endroit se situe dans la partie S2 de la protéine S toute proche d’une position très particulière près des résidus 986 et 987 qui sont des prolines à l’état naturel du virus. Les prolines ont été mutées artificiellement dans la conception des vaccins à ARNm de Pfizer et Moderna pour stabiliser la protéine complète qui en dehors de l’assemblée virale perd sa conformation active. La région spatiale autour de ces mutations très stabilisantes est supposée être une cible antigénique performante pour les anticorps neutralisants (nAb) dans la conception de ces vaccins. Bien que des anticorps neutralisants puissent être générés a priori en tout endroit bien exposé au cytoplasme, à la surface de l’assemblée supramoléculaire de la protéine S en trimère, toute mutation dans cette région fait craindre que le pouvoir du vaccin soit partiellement compromis. Si c’est effectivement le cas, ce qui n’est pas impossible, alors ce serait donc raté pour Pfizer et Moderna ainsi que pour Jansen Pharmaceuticals (autre groupe ayant développé un vaccin à ARNm). Seules ces sociétés le savent, car elles ont les données intégrales concernant l’élaboration de leurs vaccins et leur capacité à générer des anticorps en de multiples endroits du trimère S (ou pas). Nul doute possible qu’elles ont vérifié cela.
De plus, malheureusement, les anticorps générés ne sont pas forcément neutralisants, mais peuvent être également sensibilisants et déclencher les réactions d’hypersensibilisation auto-immune. En principe les autorités de santé doivent connaître ces informations avant d’autoriser un vaccin. Car si le vaccin s’avérait inutile en plus de ses effets secondaires potentiels à long terme et les milliards qu’il coûte alors le seul bénéficiaire de cette opération serait Big Pharma. De façon générale, toute mutation de surface de ces protéines, mutations qui sont les plus à même de se produire naturellement, peut potentiellement réduire l’efficacité vaccinale et leur accumulation ne peut être que délétère. Par exemple, la très forte variabilité du virus de la grippe force à recomposer le vaccin chaque année : « Les virus grippaux ont la fâcheuse tendance à muter particulièrement vite. De sorte qu’attraper la grippe une année n’immunise que rarement contre la souche qui circulera l’année suivante, car elle est suffisamment différente pour tromper notre système immunitaire. Raison pour laquelle aussi, la composition du vaccin contre la grippe dite saisonnière doit être revue chaque année. » (Sciences et Avenir). En réalité, c’est l’immunisation vaccinale qui n’est pas reconductible annuellement, car lorsque l’on attrape réellement la grippe une année il est rare d’en refaire une autre avant de très nombreuses années, voire des décennies. Le peuple français pourrait donc découvrir bientôt que l’oligarchie financière à déjà un plan de revaccination pour l’année 2022…
Une des deux autres régions de mutations observées, d’une longueur de quelques centaines de bases, est une sous-section de la région de Lyons Weiler dans le domaine N-terminal de S1. Elle a été identifiée par le Pr Montagnier et le mathématicien Jean-Claude Perez comme une région dont la composition ne semble pas en harmonie avec le reste du virus à cause de la présence concentrée de courtes séquences (20 à 30 nt). Ces séquences sont identiques ou très similaires à celles que l’on peut trouver dans des protéines essentielles du VIH, comme la protéine gp120 responsable de la pénétration du virus dans les lymphocytes T par l’intermédiaire du récepteur CD4. Le Pr Montagnier avait annoncé dans son interview d’avril que les mutations auraient une propension à apparaître dans les régions naturellement peu harmonieuses du virus, ou à des endroits qui auraient pu être manipulés.
Le second endroit de mutation correspond au site de clivage de la furine, à l’intersection des domaines S1 et S2, qui a fait également couler beaucoup d’encre et que nous traiterons dans la suite du chapitre. Ce site de clivage, entre les domaines S1 et S2, existe chez 2 des quatre coronavirus de rhume saisonniers (hCoV-HKU1 et hCoV-OC43 réputés pour leur transmission efficace chez l’homme), mais n’est pas observé chez tous les autres coronavirus connus de type SARS. Sans être nécessaire au SARS-Cov2, il semble avoir renforcé son pouvoir de pénétration. Une mutation à cet endroit est potentiellement reliée à une diminution de pathogénicité du virus ce qui serait une bonne nouvelle, mais aussi indicatif qu’un tel site à cet endroit ne serait pas ou peu naturel au virus.
Partie 2 — Critique de l’analyse de Lyons Weiler et aspects remarquables des génomes du SARS-Cov2 et des coronavirus de pangolin
Dans les parties 2, 3 et 4 de ce chapitre, nous allons regarder de près quelles sont les raisons qui font que l’on peut se demander si le SARS-Cov2 est bien d’origine naturelle. L’objection unanimement portée par la science officielle est que les anomalies génomiques relevées dans le SARS-Cov2 sont des effets de recombinaisons de segments entiers de virus et de mutations ponctuelles naturelles. Il est certain que la nature procède à de nombreuses recombinaisons de virus. Pour qu’elles aient lieu, un ou plusieurs hôtes intermédiaires doivent être infectés en même temps par au moins deux virus susceptibles de se recombiner. L’acquisition d’une fonction nouvelle (comme l’addition du site de clivage de la furine) nécessite à la fois des recombinaisons et des mutations tout cela soumis à la pression évolutive qui élimine l’immense majorité des nouveaux virus moins optimaux que ceux qui les précèdent.
C’est grâce à l’utilisation du procédé de recombinaison naturelle détourné que l’on peut fabriquer artificiellement des virus recombinés dans des cellules bactériennes (voir chapitre précédent, partie 1). Ces virus dits « recombinants » ne sont distinguables de virus naturels que par leurs structures de toute évidence hybrides, comme le branchement du domaine S1 dans un adénovirus pour en faire un vaccin. En ce qui concerne les mutations ponctuelles, leurs fréquences et leurs positions peuvent être révélatrices de manipulations possibles.
L’objection corollaire répandue, à savoir qu’une manipulation du virus aurait laissé des traces de sites de restriction, n’est pas à prendre au pied de la lettre. Comme nous l’avons vu précédemment, leur présence est tout à fait naturelle et il en existe toute une panoplie le long des génomes avec une fréquence assez élevée permettant ainsi les manipulations. Nous renvoyons le lecteur au chapitre précédent pour plus ample information. Cependant, sur ce point, il faut rappeler ici que des problèmes techniques concernant la limitation de la longueur des segments de synthèse (200 nt maximum) peut conduire dans certains cas à la nécessité d’introduire artificiellement, par PCR, des sites de restriction encadrant exactement l’endroit choisi pour la manipulation. L’introduction de ces sites va changer la composition en acides aminés autour de la séquence manipulée pouvant compromettre sa cohérence structurelle et sa fonction. Il peut donc être nécessaire de les effacer par manipulation inverse effectuée également par PCR. On évite le plus possible de faire cela pour gagner du temps et éviter de multiplier les risques d’erreurs abrogeant le succès de l’expérience.
Certains aspects du génome du SARS-Cov2 ont attiré l’attention et l’étonnement de nombre de chercheurs scientifiques qui ont émis des doutes sur l’origine naturelle du virus. Ils recouvrent les observations suivantes : (a) faible cohérence d’une grande partie du domaine S1 ; (b) manipulation suggérée du motif RBM de liaison au récepteur ACE2 ; (c) insertion du site de clivage de la furine ; (d) présence de très courts fragments du virus VIH (20 à 30 nt) ; (e) rupture d’harmonie fractale du génome du SARS-Cov2.
Les points soulevés par les chercheurs concernent principalement le domaine S1 du gène S du SARS-Cov2, qui recouvre le domaine baptisé région de Lyons Weiler (1378 nt), du nom du premier chercheur qui dès janvier 2020 a pointé une forme d’inadéquation par rapport au reste du génome. Le point (e) concernant les travaux du mathématicien Jean-Claude Perez et du Professeur Luc Montagnier qui nous révèlent une chose extraordinaire sur l’harmonie sous-jacente de la nature et l’adaptation presque parfaite du SARS-Cov2 à l’homme.
Arguments de Lyons Weiler soutenant l’hypothèse d’une recombinaison artificielle au niveau du domaine S1
C’est le chercheur universitaire Lyons Weiler qui ouvre le bal sur son site internet, le 30 janvier 2020, en exposant les données et les arguments qui le poussent à affirmer que le SARS-Cov2 pourrait être le fruit d’une expérimentation de recombinaison virale qui aurait fuité d’un laboratoire de recherche à l’Institut de virologie de Wuhan. Lyons Weiler a fondé The Institute for Pure and Applied Knowledge, USA, après avoir été Senior Research Scientist à la tête du laboratoire de bio-informatique de l’Université de Pittsburgh, de 2007 à 2014. Il est auteur de 80 articles publiés dans des revues scientifiques à comité de lecture et référencés sur PubMed.
Son argument principal réside dans l’observation que la séquence du génome du SARS-Cov2 perd une forme de cohérence sur une section contiguë d’une longueur de 1378 nucléotides (nt) soit 1.4 % de sa longueur totale. Sa mesure de cohérence se base sur la comparaison avec le virus ZC45, un coronavirus de type SARS, issu de la recherche militaire (voir chapitre 4 partie 4), le plus proche parent du SARS-Cov2 connu en janvier 2020. Si l’on compare des tronçons de quelques centaines de nucléotides entre les génomes de ZC45 et SARS-Cov2 on s’aperçoit d’une perte de niveau de corrélation (exprimé en % d’identité) sur une section d’une longueur de 1378 nt. Elle recouvre la portion N-terminale du domaine S1, plus la moitié de la portion C-terminale du domaine S1, jusqu’au motif de liaison au récepteur ACE2 de la protéine spicule du SARS-Cov2, soit 460 acides aminés. Cette section correspond à une perte de 20 % d’identité de séquence par rapport au reste des génomes des deux virus comparés.
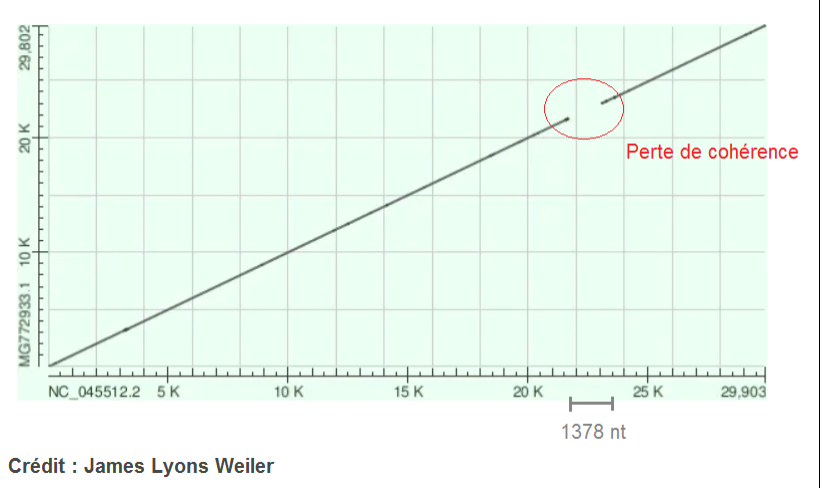
Le second argument de Lyons Weiler est que l’arbre phylogénique établissant les liens de parenté entre les virus, sur la base de leurs génomes complets, ne donne qu’une mesure d’auto-cohérence de 76 % sur le positionnement du SARS-Cov2 par rapport aux autres lignées évolutives de coronavirus. Ce résultat, établi par une équipe de recherche 100 % chinoise, est significatif et doit attirer l’attention. Il nous dit que le SARS-Cov2 ne s’inscrit pas avec la même fiabilité que les autres dans l’arbre généalogique des coronavirus. Il faut noter ce problème sans pour autant en déduire forcément que son origine n’est pas naturelle. L’arbre évolutif montre également deux autres branches avec moins de 100 % de cohérence, en particulier la branche du SARS-Cov de 2002-2003 avec 86 % de cohérence par rapport aux autres coronavirus, sans cependant être aussi bas que les 76 % du SARS-Cov2. Cela signifie que les SARS-Cov ont quelque chose de particulier par rapport aux autres virus déjà recensés au moment de l’étude et qu’il manque des branches à l’arbre. À noter que la branche du MERS n’est pas non plus auto-cohérente à 100 %.
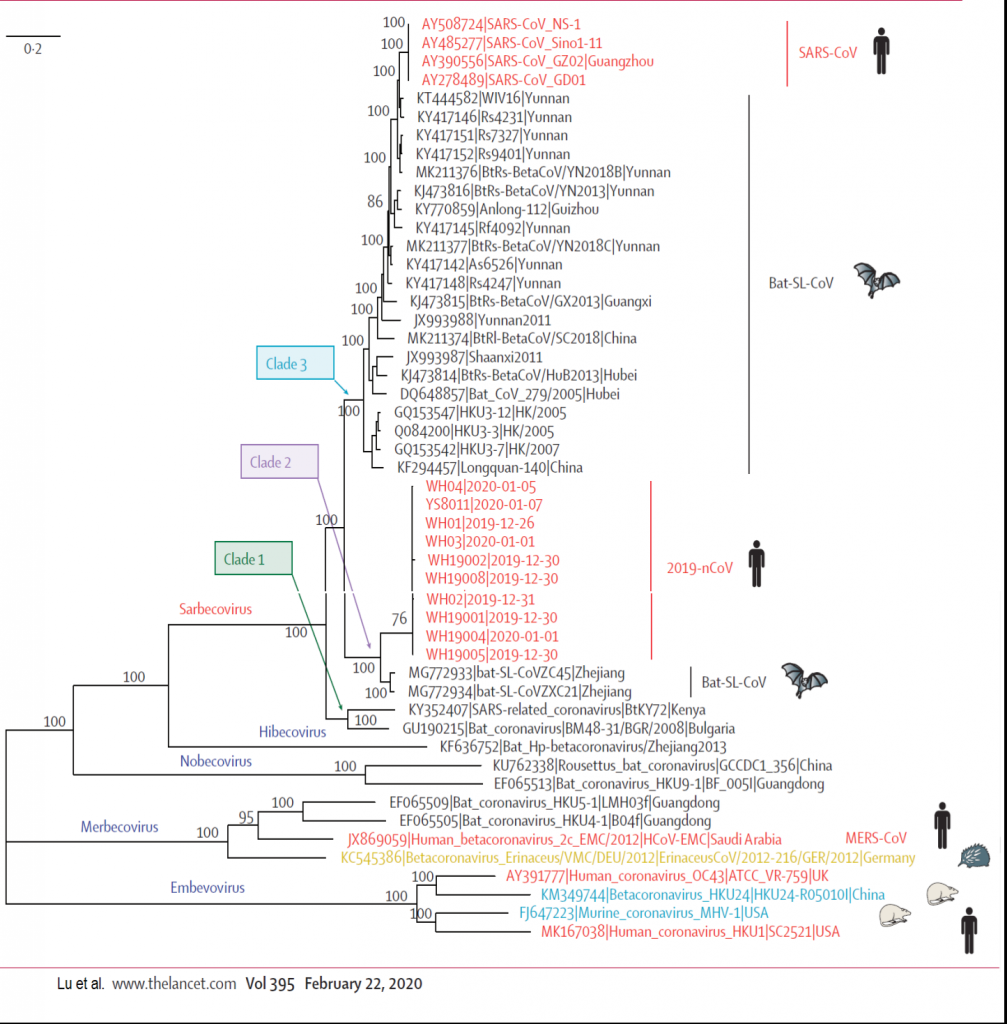
L’argument corollaire de Lyons Weiler est que la protéine spicule des coronavirus à SARS fait l’objet d’intenses recherches de manipulation de génie génétique depuis une vingtaine d’années dans le but de déterminer les facteurs moléculaires permettant le franchissement de la barrière des espèces et tenter d’établir des protocoles de création de vaccins. Dans ce but, de nombreux outils de manipulation génétique, utilisables en routine, ont été mis au point, brevetés et commercialisés, comme le plasmide p-shuttle-SN issu de la recherche chinoise. Il permet la manipulation et l’échange de la partie N-terminale de la protéine S insérée dans un adénovirus inactivé dans un but vaccinal anti SARS. Lyons Weiler est très largement critiqué pour ses positions anti-vaccin et l’Institut Pasteur de Shanghaï par l’intermédiaire de Pei Hao, un imminent chercheur et membre de l’Académie des Sciences chinoise, s’est empressé de faire paraître une lettre de réfutation référencée par PubMed, la 628e parution scientifique de sa longue carrière. Mais passons, sur les comparaisons de pedigree scientifique pour nous concentrer sur les arguments. Pei Hao avance deux arguments : (1) il est faux de dire que la séquence identifiée par Lyons Weiler est unique, car elle partage quand même environ 70 % d’identité avec les séquences équivalentes tirées des protéines S d’autre coronavirus ; (2) la similarité de cette séquence avec celle portée par le plasmide pShuttle-SN s’explique par le fait que celui-ci est fabriqué à partir du SARS-Cov.
Pei Hao a raison en ce concerne le point 2, mais son point 1 ne veut rien dire parce que cette région est très variable entre les coronavirus et donc forcément celle du SARS-Cov2 revêt un caractère d’unicité. La comparaison systématique entre les coronavirus les plus proches du SARS-Cov2, pris deux par deux, montre la divergence systématique du domaine S1 de la protéine S, en particulier de son sous-domaine N-terminal et du RBM. Les pourcentages d’identité des séquences de ces sous-unités sont systématiquement entre 5 % et 20 % inférieurs à ceux des génomes complets. Les exceptions étant les couples de virus très proches ZC45 et ZXC21 (vert) et SARS-Cov2 et RatG13 (bleu soutenu). Ces couples de virus ne présentent pas d’incohérence dans la région de Lyons Weiler et sont donc très cohérents entre eux. Les seuls virus en cohérence totale l’un par rapport à l’autre sont ZC45 et ZXC21 (vert) que l’on peut donc considérer comme des virus jumeaux. SARS-Cov2 et RaTG13 malgré une grande proximité montrent une incohérence d’identité au niveau du RBM. On note de façon générale que la partie S1-N-terminale est systématiquement plus divergente que l’ensemble du domaine S1 sauf en deux occasions (bleu soutenu) ce qui semble être une possibilité peu commune.
Lorsqu’il a fait son calcul, Lyons Weiler ne disposait pas des séquences des virus RaTG13, Pan-Cov-GD et Pan-Cov-GX. Mais il disposait du virus SARS-Cov (et même des virus RsSCH014 et Rs3367, en fait légèrement plus proches) et il aurait dû s’apercevoir que, dans une moindre mesure, l’incohérence apparaissait également entre SARS-Cov2 et SARS-Cov avec une baisse de 15 % d’identité de séquence sur la même zone. Nous avons indiqué les paires de virus pour lesquelles la baisse d’identité de séquence dans la région concernée se situe à 15 % (jaune) et à 20 % (rouge) et conduirait à l’observation faite par Lyons Weiler. Cela concerne les 2/3 des paires (15/21). Cela prouve que ce que Lyons Weiler a mesuré n’est peut-être pas une propriété particulière du SARS-Cov2, mais une propriété générale des coronavirus. Mais assurément, la mesure de Lyons Weiler montre que la divergence du domaine S1 permettrait de mesurer le lien de parenté entre les coronavirus Bat-SL-cov plus sûrement que le pourcentage d’identité globale.

De ces comparaisons, il ressort de façon remarquable que le virus Pan-Cov-GX serait en cohérence au sens de Lyons Weiler, en ce qui concerne S1, avec le virus SARS-Cov2 et RatG13 (bleu ciel). De même, Pan-Cov-GD serait en cohérence avec les virus ZC45 et ZXC21 (bleu ciel et bleu soutenu). Par contre, il n’y aurait pas de cohérence entre les virus Pan-Cov-GX et Pan-Cov-GD. Rappelons que ces deux virus proviennent de saisies de pangolins par les douanes chinoises à 18 mois d’intervalle entre juillet 2017 et janvier 2018 (Pan-Cov-GX) et mars 2019 (Pan-Cov-GD) (voire chapitre 6, partie 4). Ces mesures tendraient à montrer que les deux coronavirus ayant tué les pangolins sont distincts sur le plan de l’évolution.
Le plus remarquable est que la composition en acides aminés des RBM de Pan-Cov-GX et Pan-Cov-GD est remarquablement plus proche du RBM du SARS-Cov2 et du RatG13, aux alentours de 75 %, ou plus, alors qu’entre tous les autres coronavirus l’identité est ≤ 50 % et descend jusqu’à 31 %. Cela indique une forte similarité des RBM des virus de pangolins avec celui du SARS-Cov2 et, de façon tout à fait exorbitante, nous observons une identité de 98.6 % (un seul changement Q498H, sans impact fonctionnel potentiel marqué, sur une séquence de 72 acides aminés) entre le RBM du SARS-Cov2 et celui de Pan-Cov-GD. Nous avons décrit ce fait tout à fait étrange dans la partie 4 du chapitre 6. Nous en avions conclu que cela impliquait : soit un taux de mutation non synonyme sur cette section du génome tout à fait hallucinant (impossible à obtenir en l’absence d’un forçage comme celui décrit au chapitre 2), soit une recombinaison aléatoire favorable du RBM tout à fait exceptionnelle puisqu’elle correspondrait à un échange avec un virus déjà quasi optimal pour infecter l’être humain. Cela nous semblait impossible étant donné que le récepteur ACE2 du pangolin est très éloigné du récepteur humain (dans la partie suivante consacrée à Li-Meng Yan nous reviendrons sur ce problème). Donc, s’il y a eu recombinaison ce serait entre des virus circulant à bas bruit chez les êtres humains.
En ce qui concerne les virus ZC45 et ZXC21, la comparaison de leurs RBM avec les autres coronavirus de type SARS révèle à quel point ils sont très éloignés des autres coronavirus à SARS. Pourtant cela ne les empêche pas de potentiellement créer ce type de maladie pulmonaire chez les rats de laboratoire avec pénétration cérébrale (voir chapitre 4 partie 4).
La théorie peu convaincante du pangolin hôte intermédiaire « purificateur »
Les pourcentages d’identité du RBM, calculés entre toutes les paires de coronavirus les plus proches du SARS-Cov2, ne font que renforcer l’idée que quelque chose de tout à fait exceptionnel s’est produit, rendant très peu crédible la théorie qui tente de démontrer sans apporter de preuve que le RBM du virus Pan-Cov-GD serait le résultat d’une évolution convergente dans deux hôtes différents. Comme cette théorie n’a pas de fondement réaliste, une autre théorie, celle du pangolin comme hôte intermédiaire « purificateur » a été élaborée pour faire l’objet d’un article publié dans Science. C’est le fruit d’une collaboration sino-américaine au plus haut niveau de sérieux puisque le premier auteur Xiaojun Li est affilié au « ô combien célèbre » Los Alamos National Laboratory et le dernier auteur Feng Gao au Engineering Laboratory for AIDS Vaccine de l’University de Changchun en Chine. Dans cette théorie le RBM du virus Pan-Cov-GD aurait été acquis suite à une série de recombinaisons.
Cette théorie n’est pas prouvée formellement, car le RBM du SARS-Cov2 est très long (72 aa) et on ne peut pas trouver un sous-groupe des quelques 43 coronavirus connus de type SARS-like (c.-à-d. proche du type SARS) qui puisse permettre sa reconstruction. En effet, il faut procéder par morceaux adjoints ou superposés de séquence qui seraient complémentaires avec d’autres fragments de séquence du RBM du RaTG13. Cependant, il y existe une très grande variabilité malgré une structure sous-jacente très similaire caractérisée par une longueur souvent comparable (71-72 aa) et des acides aminés 100 % conservés dans l’évolution à des positions fixes ce qui signe l’appartenance du virus à la classe de type SARS. Par exemple, le RBM du SARS-Cov est tout à fait similaire de ce point de vue à celui du SARS-Cov2, c’est d’ailleurs pour cette raison que l’Institut Pasteur savait dès le 15 janvier que le SARS-Cov2 était un virus potentiellement très dangereux. À ce titre d’ailleurs il est étonnant de constater à quel point le RBM du virus Pan-Cov-GX partage deux sections de son RBM, de 9 et 22 aa, quasiment totalement identiques aux RaTG13 et SARS-Cov2. Cela confirme sa proximité évolutive avec ces deux virus suggérée par l’analyse de la section précédente.
Il faudrait donc faire appel à un processus de recombinaisons si multiple qu’il correspondrait à un grand nombre de mutations ponctuelles (au moins une vingtaine). D’ailleurs Xiaojun Li l’admet puisqu’il suggère des recombinaisons qui auraient eu lieu en amont et en aval du RBM favorisant l’insertion d’un RBM provenant d’un virus intermédiaire non identifié jusqu’à présent.
Les recombinaisons de virus ont lieu de façon aléatoire quand la polymérase s’arrête par accident de transcrire le code génétique du virus et que des brins d’ARNm semi-transcrits se recombinent entre eux. Dans leur immense majorité, les recombinaisons ne sont pas viables, mais lorsqu’elles le sont elles peuvent conduire à des virus radicalement différents des virus initiaux.
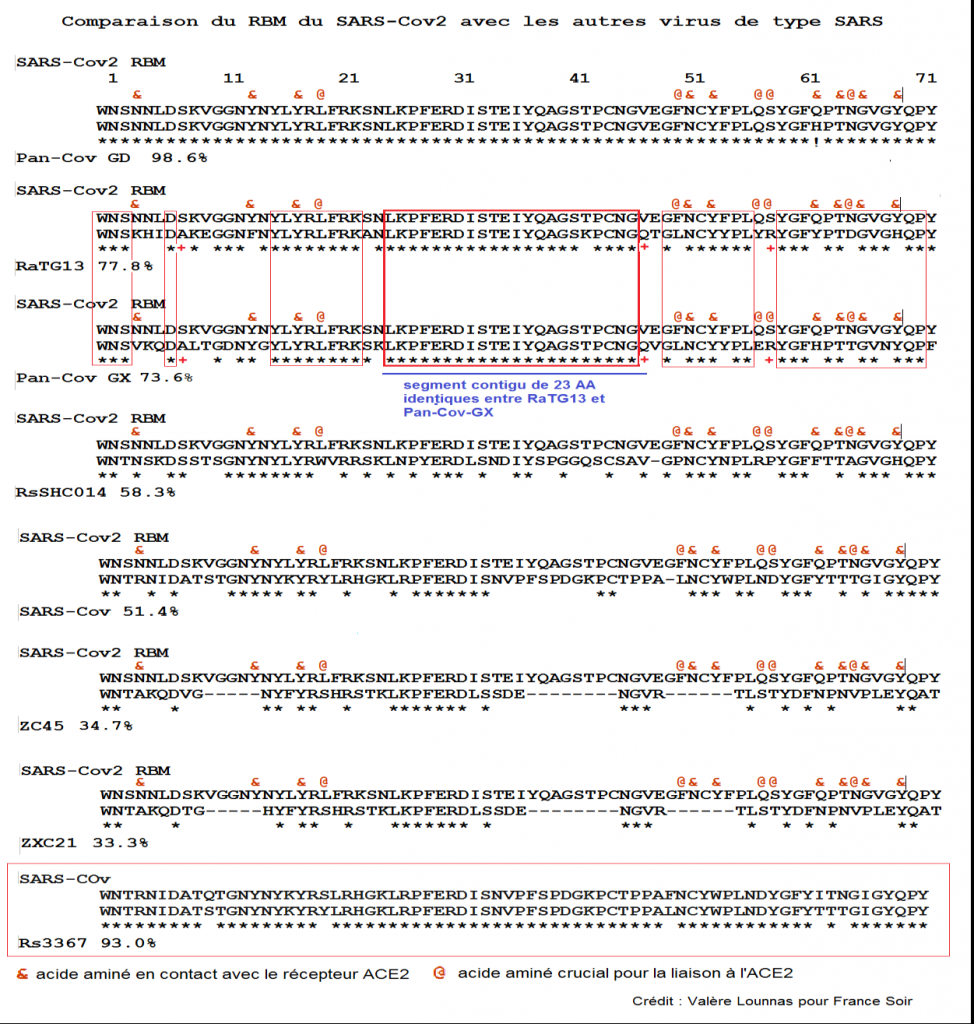
La détection des recombinaisons qui ont pu avoir lieu peut se réaliser avec un programme heuristique (empirique) RIP ou RDP3 qui détermine automatiquement les portions potentielles de génomes susceptibles de s’être échangées entre des virus connus du même type ou de types voisins. Cela se base sur la comparaison systématique de morceaux de séquence d’un virus par rapport à deux autres susceptibles de s’être recombinés. Les recombinaisons potentielles doivent être confirmées phylogénétiquement pour s’assurer qu’elles aient pu effectivement avoir se réaliser le plan spatial géographique et temporel. Toute la difficulté est là. Par exemple, il est impossible que le variant anglais de décembre 2020 soit une recombinaison d’une souche initiale de Wuhan, disparue depuis lors, avec un variant provenant d’Afrique identifié par l’équipe du Pr Raoult en septembre octobre à l’IHU-Méditerranée de Marseille. Mais les contraintes évolutives peuvent favoriser systématiquement certains types de recombinaisons qui peuvent donc se reproduire en des lieux différents. Les variants similaires créés, différent cependant en d’autres endroits de leurs séquences par rapport au premier variant repéré.
Xiaojun Li écrit cette phrase qui souligne le point faible de son article : « En comparant avec le logiciel RIP le virus Wuhan-Hu-1 (NDA premier isolat humain du SARS-Cov2) aux virus Pan_SL-CoV_GD et RaTG13, comme représentant de deux branches distinctes d’hôtes dans l’histoire de l’évolution du SARS-CoV-2 nous identifions de nombreux points de recombinaisons possibles en amont et en aval du RBM. Cela suggère que le SARS-Cov2 serait issu de recombinaisons entre les coronavirus de chauve-souris et de pangolins ».
Il ne s’agit que d’une suggestion et non d’une preuve formelle. Par ailleurs, cela suppose que les coronavirus de pangolins existent en tant que tels. Mais malheureusement tout porte à croire qu’ils ont été infectés par les contrebandiers chinois. Rappelons qu’en dehors des pangolins de Ganxji et Guangdong aucun autre pangolin, captif ou à l’état sauvage, n’a été analysé pour détecter la présence de coronavirus. Pourtant, ce serait tout à fait réalisable d’envoyer une mission effectuer des prélèvements sur les pangolins dans le Sud-est asiatique à l’instar de Shi zheng Li qui était allée chercher dans une grotte du Yunnan le virus ancêtre du SARS-Cov de 2002-2003.
Comme le rappelle à juste titre Li-Meng Yan dans son article « Une arme biologique totale », l’équipe du Dr Daszak a examiné 334 échantillons de pangolins, collectés en Malaisie et Sabah entre août 2009 et mars 2019 sans trouver la moindre trace de coronavirus. Comme Shi Zheng Li, Peter Daszack sait très bien qu’il faut aller chercher les virus là où ils peuvent se trouver. Il est directeur de l’institut non gouvernemental Eco Health Alliance (New York) et avait collaboré avec Shi Zheng Li concernant la traque du virus progéniteur de l’épidémie de 2002-2003 dans le Yunnan. Cette traque intelligente les avait conduits à l’identification entre 2011 et 2012 des virus RsSCH014 et Rs3367, parents du SARS-Cov. Avec la même rigueur logique, Peter Daszack a grandement contribué à fermer définitivement la porte à la possibilité que le pangolin puisse avoir été un hôte intermédiaire.
D’autre part, on ne voit pas très bien comment des pangolins en provenance du Vietnam en mars 2019 auraient pu développer des recombinaisons virales avec un virus extrait en 2013 d’une mine désaffectée du Yunnan, située à plus de 2000 km de distance de leur habitat sauvage et stocké en laboratoire avant d’infecter des contrebandiers. Il ne faut pas oublier que ce sont les pangolins qui sont morts, et non les contrebandiers, ce qui indique plutôt une contamination des animaux par les contrebandiers et non l’inverse. Leur décès systématique démontre que les pangolins étaient naïfs au virus. La logique des faits contredit donc fortement la conclusion de cet article. Seule la découverte d’autres coronavirus de pangolins ou d’autres espèces animales avec un pourcentage d’identité au — dessus de 97 % pourrait confirmer l’hypothèse de cet article. Mais nous en doutons fort et le peu d’empressement que la Chine met à investir dans la recherche de l’animal hôte intermédiaire semble nous donner raison.
En raison des ravages que fait le SARS-Cov2 dans les élevages de visons en Europe, et la rétro-transmission du virus des visons à l’homme, il a été proposé récemment que ces animaux puissent avoir joué un rôle intermédiaire dans l’origine de l’épidémie. Mais, a priori, cela ne semble pas une piste sérieuse. Il est impossible qu’une telle chose ait pu passer inaperçue initialement. D’ailleurs, l’annonce de cette hypothèse aurait grandement arrangé la Chine en dédouanant ses marchés aux animaux sauvages ainsi que l’Institut de Virologie de Wuhan et son laboratoire P4. Les visons certainement comme les pangolins sont très probablement les victimes collatérales du virus véhiculé par les hommes. Mais, on voit qu’ils peuvent facilement amplifier l’épidémie et on se rend compte du danger des fermes d’élevages d’animaux qui ne sont pas des animaux domestiques avec lesquels l’homme a coévolué ces derniers millénaires.
Mise au point d’un RBM optimal en laboratoire possible à partir de 2018
Avant de conclure, nous ouvrons un a parte sur la mise au point possible en laboratoire d’un RBM optimal pour infecter l’homme. Nous avons déjà largement expliqué au chapitre 3 partie 2 comment Shi Zheng Li et Ralph Baric avaient construit le premier virus Covid synthétique en branchant la protéine S du virus RsSCH014 (95,6 % d’identité avec le SARS-Cov et 80.6 % avec le SARS-Cov2) sur le SARS-Cov adapté aux souris. Ils s’étaient ensuite rendu compte, ô miracle, de la pleine capacité du virus hybride à infecter les cellules pulmonaires humaines. Shi Zheng Li qui avait identifié les virus RsSCH014 et Rs3367 (95,9 % d’identité avec le SARS-Cov) dans le Yunnan venait d’établir à quel point la transmission à l’homme était conditionnée par la protéine S. Le virus Rs3367 a été également séquencé dans le laboratoire de Shi Zheng Li et cultivé sous le nom de SL-Cov-WIV1. Ce qui n’est pas claironné sur les toits c’est que le RBM du Rs3367 est 93 % identique à celui du SARS-Cov au niveau des acides aminés (seuls 5 sur 71 aa diffèrent, 97.2 % d’identité au niveau de l’ARN) malgré les quelque 8 années d’évolution qui les séparaient, ce qui accréditait d’ailleurs l’origine du SARS-Cov dans le Yunnan à 2000 km de Canton. Le virus ayant engendré le SARS-Cov était très certainement, selon le taux de mutation établi pour les Bat-SL-Cov (environ 84/an pour l’Orf1a, voir chapitre 6 partie 3), un virus jumeau au Rs3367 partageant au moins 98 % d’identité avec lui. La paire Rs3367/SARS-Cov est cohérente au sens de Lyons Weiler ce qui indique leur très proche parenté, comme entre les virus ZC45 et ZXC21. Sans l’incohérence au niveau du RBM la paire RaTG13/SARS-Cov2 le serait également. Dans la partie suivante, nous reviendrons sur ce problème qui révèle une fois de plus que décidément quelque chose cloche étrangement à ce niveau.

Depuis 2003, une intense recherche a eu lieu en Chine pour élucider sur le plan structural le mode de liaison du RBM avec le récepteur ACE2 humain. Son aboutissement a eu lieu avec la parution le 8 août 2018 de la structure par microscopie électronique du trimère de la protéine S du SARS-Cov (virus humain non muté) lié au récepteur hACE2. Il s’agissait là d’une avancée scientifique majeure, réalisée par Wenfei Song et al., une équipe 100 % chinoise basée à Pékin (Beijing Advanced Innovation Center for Structural Biology), qui ouvrait la possibilité d’optimisation du RBM en s’appuyant sur les techniques bien établies de modélisation et de simulation de dynamique moléculaire que les chercheurs chinois maîtrisent parfaitement. Cette structure (code pdb : 6ACK) du complexe S-ACE2 a révélé l’extrême complexité de la liaison du RBM sur sa longueur de 72 acides aminés. Seulement une douzaine d’acides aminés sont en contact avec le récepteur, mais ils forment ensemble une conformation structurale qui permet de les présenter au récepteur de façon très précise. En se basant sur les séquences des RBM des virus Rs3367, RsSCH014 et RaTG13 (tous issus du laboratoire de Shi Zheng Li) on peut générer 402 hypothèses de RBM possibles qui peuvent être testées in computo en quelques jours, voire 2 ou 3 semaines au plus. Les meilleures séquences peuvent ensuite être testées dans la réalité.
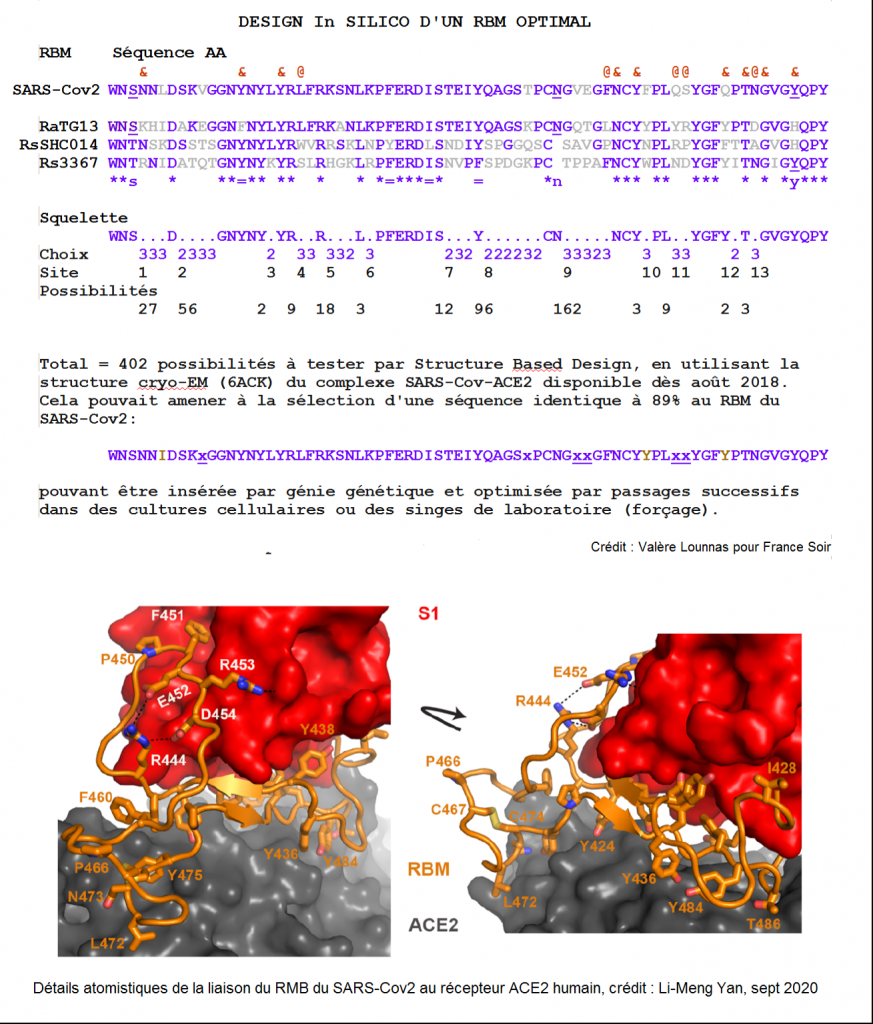
Pour résumer, nous concluons que : (a) les virus de pangolins sont des virus humains qui circulaient chez les contrebandiers chinois ; (b) entre juillet 2017 et janvier 2018 circulait chez l’homme le virus Pan-Cov-GX apparenté par son RBM au RaTG13 ; (c) en mars 2019 circulait chez l’homme un virus à 90 % identique au SARS-Cov2 et dont le RBM était 98,6 % identique à celui du SARS-Cov2 ; (c) les 90 % d’identité du Pan-Cov-GD interdisent que ce virus soit le progéniteur direct du SARS-Cov2. L’adaptation naturelle directe de son RBM à l’homme ne paraît pas une hypothèse raisonnable dans l’écosystème naturel des chauves-souris. En l’absence de l’identification d’un virus proche chez un animal intermédiaire, présent sur un marché aux animaux sauvages, elle ne peut logiquement provenir que d’une expérience GOF, par design moléculaire, suivi d’un forçage expérimental adaptatif d’un coronavirus proche du RaTG13 chez un primate de laboratoire. Évidemment, la logique n’est pas une preuve, mais une indication vers où chercher. Une expérience similaire aurait pu conduire au Pan-Cov-GX. Un ou plusieurs virus, portant un RBM identique à celui du SARS-Cov2, auraient pu déjà circuler à bas bruit dans la population de Wuhan depuis le début de l’année 2019.
Finalement, même si les conclusions que tire Lyons Weiler d’une analyse incomplète sont erronées ou du moins trop hâtives, l’extension systématique de son approche met en exergue une incohérence d’identité de séquence, semblable à celle qu’il révèle, entre la plupart des coronavirus à SARS. Cela s’expliquerait par la très grande pression évolutive sur ce gène, exarcerbée au moment du franchissement de la barrière des espèces. Cependant, en zoomant sur le RBM on se rend compte de l’existence d’autres problèmes de cohérence troublants entre le SARS-Cov2 et les deux virus les plus proches que sont le RaTG13 et le Pan-Cov-GD. Nous remercions Lyons Weiler et les personnes comme lui qui permettent d’entamer une discussion approfondie sur la question de l’origine du SARS-Cov2. Il semble exclu définitivement que les malheureux pangolins en voie de disparition soient des hôtes intermédiaires du SARS-Cov2. Ils sont par contre les témoins post-mortem que des virus à SARS circulaient en Chine entre 2017 et 2019.
En cas d’erreur flagrante de notre part, nous invitons tout chercheur confirmé en virologie à envoyer à FranceSoir une contradiction, ou une correction du raisonnement, qui sera publiée à condition évidemment qu’elle ne soit pas juste une opinion relayée, mais étayée scientifiquement par le calcul.
⚠ Les points de vue exprimés dans l’article ne sont pas nécessairement partagés par les (autres) auteurs et contributeurs du site Nouveau Monde.


















