
23/05/2023 (2022-02-22)
[Source : cv19.fr 4 février 2022]
[Mise à jour : le site cv19.fr n’est plus fonctionnel, mais on peut encore retrouver au moins une partie de son contenu sur le site Internet archive.org :
https://web.archive.org/web/20230000000000*/https://cv19.fr
avec par exemple sa copie trouvée en date du 27 juillet 2022 :
https://web.archive.org/web/20220727021211/https://cv19.fr/]
Stefan Lanka, en collaboration avec un mathématicien anonyme, vient de rendre publiques ses recherches sur l’analyse du génome du SARS-CoV-2 et des techniques et méthodes questionnables utilisées par les virologues.
C’est l’un des derniers éléments qu’il manquait pour complètement réfuter, méthodiquement, toutes allégations d’un nouveau virus contagieux responsable d’une nouvelle maladie tel que décrit et accepté actuellement.
Pour résumer simplement cette étude assez technique, la publication à l’origine du premier génome du SARS-CoV-2 n’est pas reproductible, car les données fournies ne permettent pas d’aboutir aux mêmes résultats.
Il est également démontré que les données publiées ont été manipulées (dans le sens « travaillées ») et qu’elles ne correspondent pas à ce qu’elles devraient normalement représenter tel que revendiqué.
Ce simple article, sur la base de différentes études virologiques et à l’aide de plusieurs outils bio-informatiques, permet de remettre en cause l’ensemble du bien-fondé du domaine de la génétique appliquée à la virologie moderne, et donc du SARS-CoV-2, le virus présumé responsable de la maladie Covid-19, dont la réalité repose presque exclusivement sur ces méthodes et techniques.
Vous pouvez retrouver l’étude complète au format PDF en anglais ci-dessous ou traduite en français plus bas.
Voir aussi les tableaux et figures :
Analyse structurelle des données de séquençage en virologie
Une approche élémentaire à partir de l’exemple du SARS-CoV-2
Auteur
Par un mathématicien de Hambourg, qui souhaite rester anonyme.
Abstract
Le séquençage méta-transcriptomique de novo ou le séquençage du génome entier sont des méthodes acceptées en virologie pour la détection de prétendus virus pathogènes. Dans ce processus, aucune particule virale (virion) n’est détectée et, au sens du mot isolement, isolée et caractérisée biochimiquement. Dans le cas du SARS-CoV-2, l’ARN total est souvent extrait d’échantillons de patients (par exemple : liquide de lavage broncho-alvéolaire [LBA] ou écouvillons de gorge) et séquencé. Notamment, il n’y a aucune preuve que les fragments d’ARN utilisés pour le calcul des séquences du génome viral soient d’origine virale.
Nous avons donc examiné la publication « A new coronavirus associated with human respiratory disease in China » [1] et les données de séquence publiées associées avec le bioprojet ID PRJNA603194 daté du 27/01/2020 pour la proposition de séquence génétique originale du SARS-CoV-2 (GenBank : MN908947.3). Une répétition de l’assemblage de novo avec Megahit (v.1.2.9) a montré que les résultats publiés ne pouvaient pas être reproduits. Nous avons peut-être détecté des acides ribonucléiques (ribosomaux) d’origine humaine, contrairement à ce qui a été rapporté dans [1]. Une analyse plus poussée a fourni des preuves d’une possible amplification non spécifique des lectures pendant la confirmation par PCR et la détermination des terminaisons génomiques non associées au SARS-CoV-2 (MN908947.3).
Enfin, nous avons réalisé des assemblages de référence avec des séquences génomiques supplémentaires telles que le SARS-CoV, le virus de l’immunodéficience humaine, le virus de l’hépatite delta, le virus de la rougeole, le virus Zika, le virus Ebola ou le virus de Marburg, afin d’étudier la similarité structurelle des données de séquence actuelles avec les séquences respectives. Nous avons obtenu des indications préliminaires selon lesquelles certaines des séquences du génome viral que nous avons étudiées dans le présent travail pourraient être obtenues à partir de l’ARN d’échantillons humains insoupçonnés.
Mots-clés
SARS-CoV-2, COVID-19, Virus, assemblage de novo, séquençage du génome entier, WGS, bioinformatique, PCR, SARS-CoV, Bat SARS-CoV, virus de l’immunodéficience humaine, VIH, virus de l’hépatite, virus de la rougeole, virus Zika, virus Ebola, virus de Marburg.
Introduction
Pour construire des séquences génomiques virales, les acides nucléiques (ARN ou ADN) sont isolés à partir de diverses sources d’acides nucléiques telles que le liquide de lavage broncho-alvéolaire (LBA) [1, 2], les écouvillons nasopharyngés [3, 4, 5, 6, 12, 13], les composants de culture cellulaire ou les surnageants de culture cellulaire [2, 11, 12, 13, 14, 16], ainsi qu’à partir d’échantillons humains [8, 9, 10, 16] et animaux [7, 15], puis séquencés. Dans ce processus, les acides nucléiques obtenus ne proviennent pas exclusivement de particules (de virus) préalablement isolées, c’est-à-dire séparées de tout le reste, mais souvent de l’échantillon entier. Ainsi, l’origine des fragments d’acide nucléique utilisés pour calculer les séquences génomiques n’est a priori pas claire.
Dans le cas des acides ribonucléiques (ARN), ceux-ci sont d’abord transcrits en ADNc à l’aide d’une ADN polymérase ARN-dépendante. L’ADN ou l’ADNc est ensuite fragmenté à l’aide d’enzymes et amplifié par réaction en chaîne par polymérase (PCR) avant que le séquençage proprement dit, c’est-à-dire la détermination de la séquence nucléotidique des courts fragments d’ADN ou d’ADNc, n’ait lieu. Lors de l’amplification, outre des séquences d’amorces aléatoires (hexamères aléatoires), des séquences d’amorces hautement spécifiques sont également utilisées en fonction des génomes de référence ou des génomes cibles considérés [par exemple : 1, 3, 4, 5, 6, 7, 8, 17, 18]. Enfin, les données de séquence ainsi obtenues sont traitées à l’aide d’algorithmes bioinformatiques.
Deux méthodes courantes pour déterminer les séquences du génome viral sont l’assemblage méta-transcriptomique de novo [1, 12] et le séquençage du génome entier (whole genome sequencing) [3, 4, 5, 6, 17, 18]. Alors que l’assemblage méta-transcriptomique de novo n’utilise souvent aucune séquence de référence ou seulement des séquences de référence en aval, le séquençage du génome entier utilise un grand nombre de séquences d’amorces spécifiques, dont certaines couvrent déjà ensemble 4 à 17 % du génome cible [1, 17]. Pour l’amplification de l’ADNc, 35 à 45 cycles sont souvent utilisés [1, 6, 17].
Dans le cas du SARS-CoV-2 (GenBank : MN908947.3) [1], la proposition de séquence du génome viral a été calculée par assemblage méta-transcriptomique de novo de l’ARN total provenant du LBA (lavage broncho-alvéolaire) d’un patient de Wuhan, en Chine. Les assembleurs Megahit (v.1.1.3) et Trinity (v.2.5.1) ont été utilisés pour assembler les contigs. Megahit a généré un total de 384 096 (200 nt – 30,474 nt) et Trinity a calculé 1 329 960 (201 nt – 11,760 nt) contigs. Les grandes différences entre les deux assemblages sont notables. Selon [1], le plus long contig assemblé avec Megahit présentait une grande similarité nucléotidique (89,1 %) avec le génome de la chauve-souris SL-CoVZC45 (GenBank : MG772933) et a été utilisé pour concevoir des amorces pour la confirmation par PCR et les terminaisons du génome.
L’organisation du génome viral a été déterminée par alignement de séquences sur deux espèces représentatives du genre Betacoronavirus, un coronavirus associé à l’homme (SARS-CoV Tor 2, GenBank : AY274119) et un coronavirus associé aux chauves-souris (bat SL-CoVZC45, GenBank : MG772933).
Aucune particule virale pathogène associée de manière unique à la séquence MN908947.3 n’a été identifiée et caractérisée biochimiquement à partir de l’échantillon du patient. Au contraire, l’ARN total a été extrait et traité à partir du LBA d’un patient. Il n’y a pas de preuve que seuls des acides nucléiques viraux ont été utilisés pour construire le génome viral revendiqué pour le SARS-CoV-2. De plus, en ce qui concerne la construction du brin de génome viral revendiqué, aucun résultat d’éventuelles expériences témoins n’a été publié. Ceci est également vrai pour toutes les autres séquences de référence considérées dans le présent travail. Dans le cas du SARS-CoV-2, un contrôle évident serait que le génome viral revendiqué ne puisse pas être assemblé à partir de sources d’ARN non suspectées d’origine humaine, ou même d’autres origines.
Dans la présente publication, nous avons étudié la reproductibilité des assemblages de novo en utilisant les données de séquence originales publiées pour le travail original sur le coronavirus SARS-CoV-2 [1]. Nous avons également étudié la similarité structurelle des données de séquence actuelles avec d’autres séquences virales de référence accessibles au public pour le SARS-CoV (chauve-souris) [1, 7, 13, 14], le virus de l’immunodéficience humaine [8], le virus de l’hépatite delta [9], le virus de la rougeole [11, 12], le virus Zika [10], le virus Ebola [15] et le virus de Marburg [16] (Tableaux et Figures : Tableau 3). A cette fin, nous présentons ici un protocole bioinformatique simple. Pour valider nos résultats, nous avons également considéré des séquences génomiques générées de manière aléatoire et fictive afin d’exclure le caractère purement aléatoire de nos résultats.
Section principale
Reconstitution de l’assemblage de novo des données de séquence publiées
Pour répéter l’assemblage de novo, nous avons téléchargé les données de séquence originales (SRR10971381) du 27/01/2020 au 11/30/2021 en utilisant les outils SRA [19] à partir d’Internet. Pour préparer les lectures en paires pour l’étape d’assemblage avec Megahit (v.1.2.9) [20], nous avons utilisé le préprocesseur FASTQ fastp (v.0.23.1) [21]. Après avoir filtré les lectures en paires, 26 108 482 des 56 565 928 lectures initiales sont restées, avec une longueur d’environ 150 pb. Une grande partie des séquences, vraisemblablement une majorité de celles d’origine humaine, ont été écrasées par les auteurs avec « N » pour inconnu et donc filtrées par fastp. Ceci doit être considéré comme un problème au sens de la scientificité, puisque toutes les étapes ne peuvent pas être retracées ou reproduites. Pour la génération élaborée de contigs à partir des lectures de séquences courtes restantes, nous avons utilisé Megahit (v.1.2.9) en utilisant les paramètres par défaut.
Nous avons obtenu 28 459 (200 nt – 29 802 nt) contigs, soit beaucoup moins que ce qui est décrit dans [1]. Contrairement aux représentations de [1], le contig le plus long que nous avons assemblé ne comprenait que 29 802 nt, soit 672 nt de moins que le contig le plus long avec 30 474 nt, qui selon [1] comprenait presque tout le génome viral. Notre contig le plus long a montré une correspondance parfaite avec la séquence MN908947.3 à une longueur de 29 801 nt (Tableaux et Figures, Tableaux 1, 2). Nous n’avons donc pas pu reproduire le contig le plus long de 30 474 nt, qui est si important pour la vérification scientifique. Par conséquent, les données de séquence publiées ne peuvent pas être les lectures originales utilisées pour l’assemblage.
Après avoir assemblé les contigs, nous avons déterminé la richesse de couverture respective en faisant correspondre les séquences courtes aux 28 459 contigs déterminés à l’aide de Bowtie2 (v.2.4.4) [22]. Nous avons ensuite fait correspondre les 50 contigs ayant la plus grande abondance de couverture et les 50 contigs les plus longs à la base de données de nucléotides (Blastn) le 12/05/2021 et le 12/20/2021, respectivement. Les résultats détaillés des requêtes se trouvent dans les tableaux et figures : Tableaux 1, 2.
Une comparaison de nos résultats (Tables and Figures: Table 1) avec ceux de [1, Supplementary Table 1. The top 50 abundant assembled contigs generated using the Megahit program.] montre des différences remarquables. Dans ce qui suit, les ID de contigs de [1] sont précédés de « 1_ » pour mieux les distinguer de nos ID de contigs. En général, on peut dire que les résultats de nos requêtes concernant les numéros d’accession ne correspondent pas exactement à ceux de [1]. En ce qui concerne les descriptions des sujets, nous avons observé une bonne correspondance pour la plupart. De plus, à l’exception du contig le plus long (1_k141_275316), nos contigs se sont avérés plus longs et ont eu tendance à avoir une couverture plus riche. Le cas est clair pour le contig 1_k141_179411 comparé au contig k141_12253. Le premier a une longueur de 2 733 nt, tandis que le second a une longueur de 5 414 nt. Cela fournit la première indication possible que l’amplification non spécifique de lectures de séquences non associées au SARS-CoV-2 s’est produite pendant la confirmation par PCR avec des amorces construites pour MN908947.3 à partir de 1_k141_275316 (30,474 nt).
À ce stade, le contig avec l’identification k141_27232, auquel 1 407 705 séquences sont associées, et donc environ 5 % des 26 108 482 séquences restantes, doit être discuté en détail. L’alignement avec la base de données de nucléotides le 05/12/2021 a montré une correspondance élevée (98,85 %) avec « Homo sapiens RNA, 45S pre-ribosomal N4 (RNA45SN4), ribosomal RNA » (GenBank : NR_146117.1, daté du 04/07/2020). Cette observation contredit l’affirmation de [1] selon laquelle la déplétion de l’ARN ribosomal a été effectuée et les lectures de séquences humaines ont été filtrées à l’aide du génome de référence humain (version 32 humaine, GRCh38.p13). Il convient de noter que la séquence NR_146117.1 n’a été publiée qu’après la publication de la bibliothèque de séquences SRR10971381 considérée ici.
Cette observation souligne la difficulté de déterminer a priori l’origine exacte des fragments individuels d’acide nucléique utilisés pour construire les séquences génomiques virales revendiquées.
Analyse de la structure des séquences basée sur les références
Nous avons d’abord mappé les lectures en paires (2×151 pb) avec BBMap [23] aux séquences de référence que nous avons considérées (Tableaux et Figures : Tableau 3) en utilisant des paramètres relativement peu spécifiques. Nous avons ensuite fait varier la longueur minimale (M1) et l’identité (nucléotidique) minimale (M2) avec reformat.sh pour obtenir des sous-ensembles correspondants des séquences précédemment cartographiées avec une qualité appropriée. L’augmentation de la longueur minimale M1 ou de l’identité nucléotidique minimale M2 augmente ainsi la significativité de la cartographie respective. Ensuite, nous avons formé des séquences consensus avec les sous-ensembles respectifs de qualité sélectionnée par rapport à la référence sélectionnée. Nous avons attribué la valeur « N » (inconnu) à toutes les bases dont la qualité est inférieure à 20. Une qualité de 20 signifie un taux d’erreur de 1 % par nucléotide, ce qui peut être considéré comme suffisant dans le contexte de nos analyses. Enfin, l’évaluation de la concordance entre les séquences de référence et les séquences consensus a été réalisée à l’aide de BWA [24], Samtools [25] et Tablet [26]. La paire ordonnée (M1 ; M2) = (37 ; 0,6) a été juste choisie pour donner des taux d’erreur F1 et F2, respectivement, de moins de 10 % pour la référence LC312715.1. Les résultats de tous les calculs effectués sont présentés dans les tableaux et les figures : Tableau 4. Les calculs montrent la signification la plus élevée pour le choix de la paire ordonnée (37 ; 0,6), ce qui peut être vu par les taux d’erreur les plus élevés dans chaque cas. Une signification comparable est fournie par les paires ordonnées (47 ; 0,50) et (25 ; 0,62). Alors que les séquences génomiques associées aux coronavirus présentent des taux d’erreur approximativement supérieurs à 10 % pour toutes les paires ordonnées considérées (M1 ; M2), les taux d’erreur des deux séquences LC312715.1 (VIH) et NC_001653.2 (Hépatite delta) sont inférieurs à 10 % et diminuent encore pour les paires ordonnées (32 ; 0,60) et (30 ; 0,60). La séquence MG772933_short est principalement constituée de la partie qui n’est pas recouvrable par les lectures associées au SARS-CoV-2 (voir Tableaux et Figures : Figure 3). Là encore, aucune amélioration n’a pu être obtenue en réduisant les valeurs de M1 et M2. Les taux d’erreur des séquences NC_039345.1 (virus Ebola), NC_024781.1 (virus Marburg), AF266291.1 et KJ410048.1 (virus de la rougeole) sont nettement plus élevés que ceux des séquences LC312715.1 et NC_001653.2. Alors que les séquences d’acides nucléiques utilisées pour calculer les premiers génomes ont été propagées dans des cellules Vero, les séquences d’acides nucléiques utilisées pour LC312715.1 et NC_001653.2 proviennent directement d’échantillons d’origine humaine (Tableaux et Figures : Tableau 3). Par conséquent, la question se pose de savoir si ce résultat est dû à des différences structurelles des sources d’acides nucléiques respectives ou aux protocoles de séquençage respectifs utilisés. Par exemple, la transcriptase inverse utilisée pour convertir l’ARN en ADNc ou les séquences d’amorces utilisées pour l’amplification ainsi que les cycles d’amplification pourraient éventuellement entraîner des différences dans les bibliothèques de séquences obtenues.
Les taux d’erreur F1 et F2 les plus élevés sont affichés par les séquences génomiques fictives générées aléatoirement rnd_uniform, rnd_wuhan, rnd_wh_mk_1 et rnd_wh_mk_2, de sorte que les résultats trouvés ici ne sont pas purement aléatoires.
Analyse graphique des distributions de couverture et des longueurs de lecture
Après avoir observé la possibilité de former des séquences consensus de haute qualité par rapport à certaines séquences de référence, nous avons analysé la distribution de la couverture des lectures de courtes séquences associées (Tableaux et Figures : Figures 1-22) et la distribution des longueurs de lecture (Tableaux et Figures : Figures 23-25). Pour ce faire, nous avons préalablement mappé les lectures de séquences courtes à leurs séquences de référence respectives en utilisant BBMap, [(M1 ; M2) = (37 ; 0,60)]. En plus des séquences courtes, nous avons également mappé les 26 paires d’amorces [1, Supplementary Table 8. PCR primers used in this study.] pour le séquençage du génome entier du SARS-CoV-2 (GenBank : MN908947.3) aux génomes de référence considérés. L’analyse subséquente a été effectuée via Tablet et le tableur Excel.
Tout d’abord, nous considérons la référence générée aléatoirement rnd_uniform. Des observations comparables s’appliquent aux génomes de référence générés aléatoirement rnd_wuhan, rnd_wh_mk_1 et rnd_wh_mk_2 (Tableaux et Figures : Figures 14-16).

a) rnd_uniform_reads cartographié à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60).
b) rnd_uniform_primer cartographié à l’aide de BBMap.
c) La couverture distribuée exponentielle a été générée par simulation stochastique à l’aide de la méthode d’inversion.
d) Les 26 paires d’amorces ([1, Tableau supplémentaire 8. Amorces PCR utilisées dans cette étude]) sont réparties de manière inégale sur l’ensemble du génome de référence. Les positions des amorces ne sont que faiblement corrélées avec les zones de couverture nucléotidique élevée, chacune ne comprenant que quelques nucléotides.
e) La distribution des rnd_uniform_reads semble largement aléatoire. La variance de la distribution exponentielle considérée correspond bien à la variance empirique ajustée.
La couverture (rnd_uniform_reads) varie de manière aléatoire et relativement homogène sur toutes les positions nucléotidiques. La structure est comparable à celle de la couverture générée aléatoirement (couverture distribuée exponentielle), bien que la variance semble un peu plus faible. À quelques positions nucléotidiques isolées, la couverture présente une couverture élevée par rapport à la moyenne, mais chacune d’entre elles ne couvre que quelques régions nucléotidiques contiguës. Une corrélation avec les positions des amorces n’est que faiblement prononcée. La couverture d’apparence purement aléatoire avec les lectures de séquences courtes est corrélée avec une séquence consensus mappable non continue et un taux d’erreur F1 élevé de 38,60 %. Ainsi, la structure aléatoire (interne) des nucléotides de la séquence de référence simulée stochastiquement « rnd_uniform » est relativement absente des données de séquence examinées ici.
Par contraste, nous considérons maintenant le génome de référence du SARS-CoV-2 (GenBank : MN908947.3).

a) MN908947_reads mappé avec Bowtie2 en utilisant les paramètres par défaut.
b) MN908947_primer cartographié à l’aide de BBMap.
c) Les quantiles ont été déterminés à partir de EN et VARN sous l’hypothèse de distribution d’une distribution binomiale.
d) Les 26 paires d’amorces ([1], Tableau supplémentaire 8. Amorces PCR utilisées dans cette étude.) sont réparties uniformément sur l’ensemble du génome de référence. Les positions des amorces sont en corrélation avec les zones de couverture nucléotidique élevée.
Contrairement à la figure 13, la distribution de la couverture présente davantage un modèle en forme de vague avec des couvertures de nucléotides régulières et significativement plus importantes. Les 26 paires d’amorces sont réparties uniformément sur toutes les positions nucléotidiques de la séquence de référence. Les positions d’amorce sont souvent situées près des positions nucléotidiques avec une couverture nucléotidique élevée par rapport à la moyenne. Cela indique que toutes les parties du génome de référence n’ont pas été amplifiées de manière égale. En supposant que les 29 903 positions nucléotidiques ont la même probabilité d’apparaître dans les lectures associées au SARS-CoV-2, la couverture de chaque position nucléotidique devrait se situer entre les deux lignes avec une probabilité de 99,5 % (en supposant une distribution binomiale). Ce n’est pas le cas pour environ 90 % des positions nucléotidiques. A priori, on pourrait s’attendre à ce que si une quantité suffisante d’ARN viral est présente dans l’échantillon et que suffisamment de morceaux de séquence sont lus, on obtienne une couverture homogène des nucléotides au sein du génome viral.
Le graphique suivant permet d’étudier les distributions des longueurs de lecture des références que nous venons de considérer (rnd_uniform et MN908947.3)

a) — f) Cartographie à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60).
Analyse dans Excel.
La figure 23e) montre la distribution des longueurs de lecture dans le cas de la référence « rnd_uniform ». La longueur moyenne des lectures est de 41,96 nt, à peine à droite du maximum de la distribution. En comparaison, la distribution pour la référence MN908947.3, Figure 23a) montre une région proéminente (aléatoire) similaire à la Figure 23e) et une région distincte avec des lectures d’environ 150 nt de longueur. La longueur moyenne des lectures est supérieure à 110 nt. Toutes les séquences de référence avec une distribution comparable et donc plutôt aléatoire des longueurs de lecture comme dans la référence « rnd_uniform » simulée de manière stochastique (Tableaux et Figures : Figure 23 d), f) ; Figure 24 d), e), f) ; Figure 25a) — c)) présentent également des taux d’erreur élevés F1 et F2 (Tableaux et Figures : Tableau 4).
Cette constatation est mise en évidence par l’analyse suivante. Afin de mieux comprendre la structure interne des quelque 56 millions de séquences publiées, nous avons considéré la condition supplémentaire maxlength=100 pour la séquence MN908947.3 lors de la formation des sous-ensembles après cartographie avec BBMap en plus de M1 et M2.
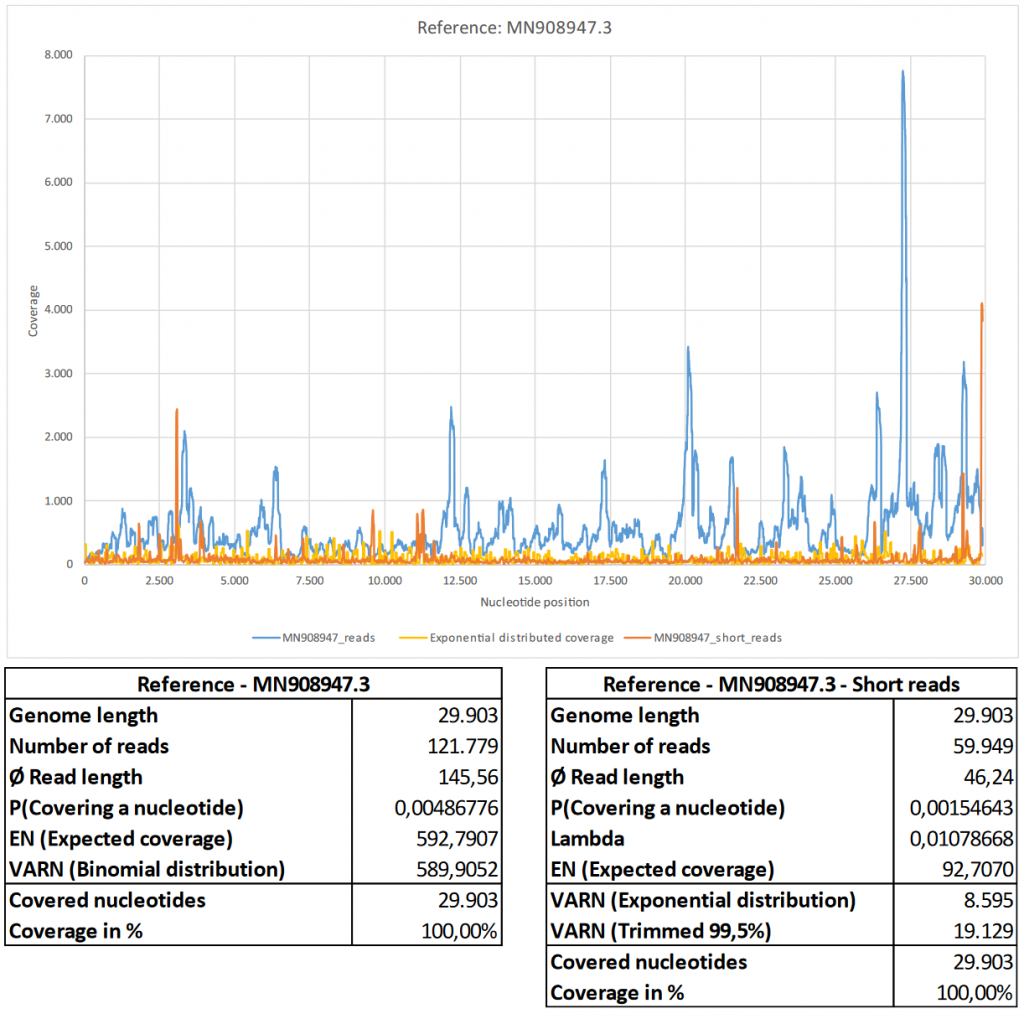
a) MN908947_reads cartographié avec Bowtie2 en utilisant les paramètres par défaut.
b) MN908947_short_reads cartographié avec BBMap, (M1 ; M2) = (37 [max. 100) ; 0,60).
c) La couverture distribuée exponentielle a été générée par simulation stochastique en utilisant la méthode d’inversion. La distribution de la couverture MN908947_short_reads présente un modèle plus aléatoire, mais sa variance ajustée est plus élevée. Ceci est principalement dû aux quelques fluctuations de la distribution de la couverture.
En excluant toutes les séquences mappables de plus de 100 nucléotides, on a essentiellement éliminé les quelque 120 000 lectures associées au SARS-CoV-2. La distribution de la couverture des courtes séquences restantes semble maintenant aléatoire, de façon analogue à la figure 13. Là encore, cela correspond aux taux d’erreur élevés de R1 (29,90 %) et R2 (29,96 %). Cela indique qu’aucune structure significative de la référence MN908947.3 n’est incluse dans les séquences publiées, à l’exception des quelque 120 000 (tableaux et figures. Tableau 1) lectures courtes associées.
Avant d’entrer dans le détail de certains des génomes de référence que nous avons examinés, nous aimerions d’abord examiner la couverture de deux autres contigs : k141_12253 et k141_20796. Alors que le contig identifié comme k141_12253 est caractérisé par une couverture relativement élevée, k141_20796 fait partie des trois plus longs contigs calculés.

a) k141_12253_reads mappé avec Bowtie2 en utilisant les paramètres par défaut.
b) k141_12253_primer cartographié avec BBMap.
Le contig k141_12253 présente une grande similarité avec la bactérie Leptotrichia (GenBank : CP012410.1). Sur les 52 séquences d’amorces publiées, 38 ont pu être cartographiées sur la référence k141_12253 avec un taux d’erreur relativement élevé de 37,30 %. La distribution de la couverture s’avère être extrêmement inhomogène et montre, surtout dans les 500 premiers nucléotides, une couverture nucléotidique extrêmement élevée par rapport à la moyenne. Les zones présentant une couverture élevée sont en corrélation avec les positions d’amorce déterminées. Cela pourrait indiquer que des lectures non exclusivement associées au SARS-CoV-2 ont été amplifiées en grande quantité. Compte tenu du taux d’erreur relativement élevé de 37,30 %, cela impliquerait une amplification relativement non spécifique. Ainsi, la question se pose de savoir si les lectures obtenues par l’amplification de l’ADNc avec les séquences d’amorce spécifiques étaient déjà présentes dans l’échantillon initial ou ont été générées par la procédure elle-même.
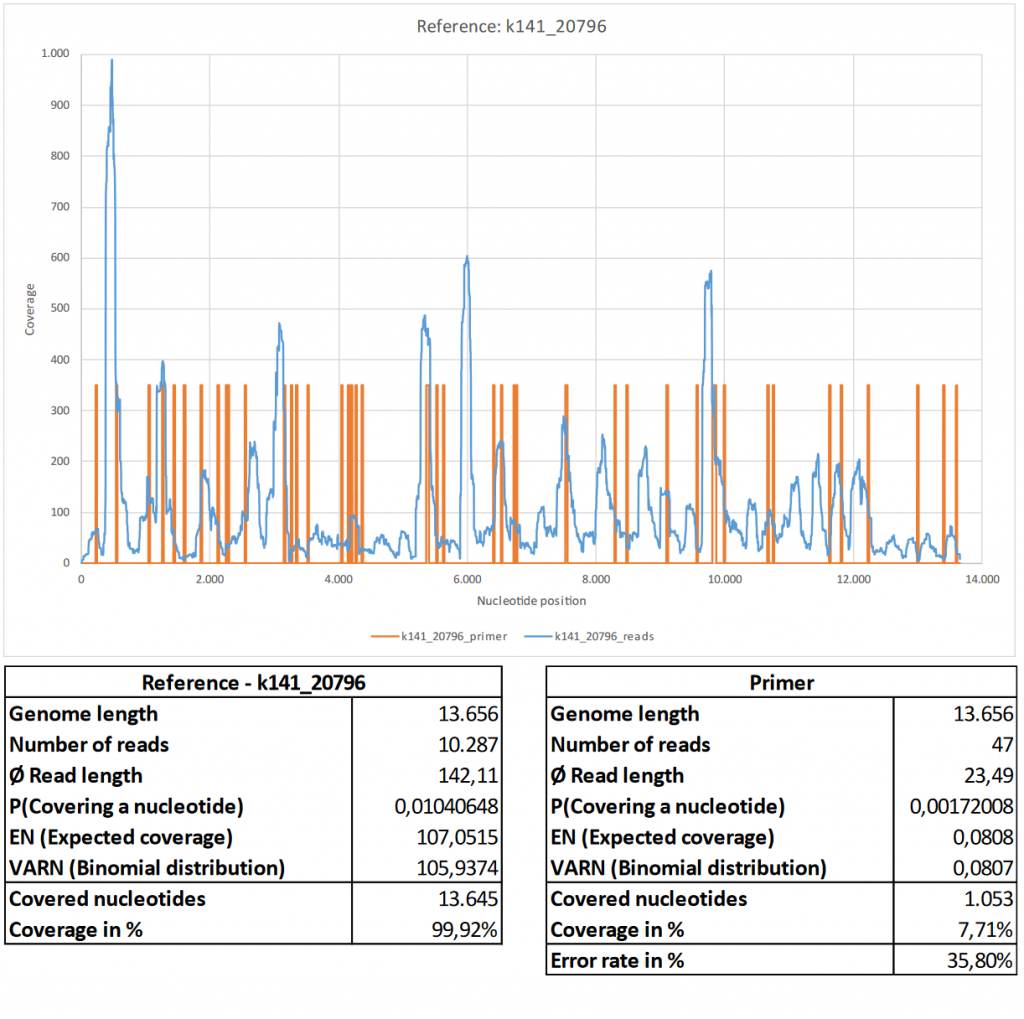
a) k141_20796_reads mappé avec Bowtie2 en utilisant les paramètres par défaut.
b) k141_20796_primer cartographié avec BBMap.
Le contig k141_20796, qui a une correspondance élevée avec la bactérie Veillonella parvula (GenBank : LR778174.1), montre une couverture plus faible avec des lectures associées par rapport au contig avec l’identification k141_12253. La structure de la couverture nucléotidique est similaire à celle du SARS-CoV-2 (GenBank : MN908947.3). Notamment, la couverture est à nouveau inhomogène, ce qui indique une amplification inégale. En raison de la longueur nucléotidique plus élevée, 47 des 52 séquences d’amorces publiées ont pu être cartographiées sur le contig de référence avec un taux d’erreur moyen de 35,80 %. Encore une fois, les positions des amorces sont bien corrélées avec les zones de couverture nucléotidique élevée. Cela pourrait à nouveau indiquer une amplification non spécifique de séquences non associées au SARS-CoV-2 (GenBank : MN908947.3).
Dans la présente section, nous examinerons plus en détail les séquences de référence « Virus de l’immunodéficience humaine 1 » (GenBank : LC312715.1) et « Virus de la rougeole de génotype D8 souche MVi/Muenchen » (GenBank : KJ410048.1). Toutes les autres figures se trouvent dans les documents supplémentaires (Tableaux et Figures : Figures 1-22 et Figures 23-25).
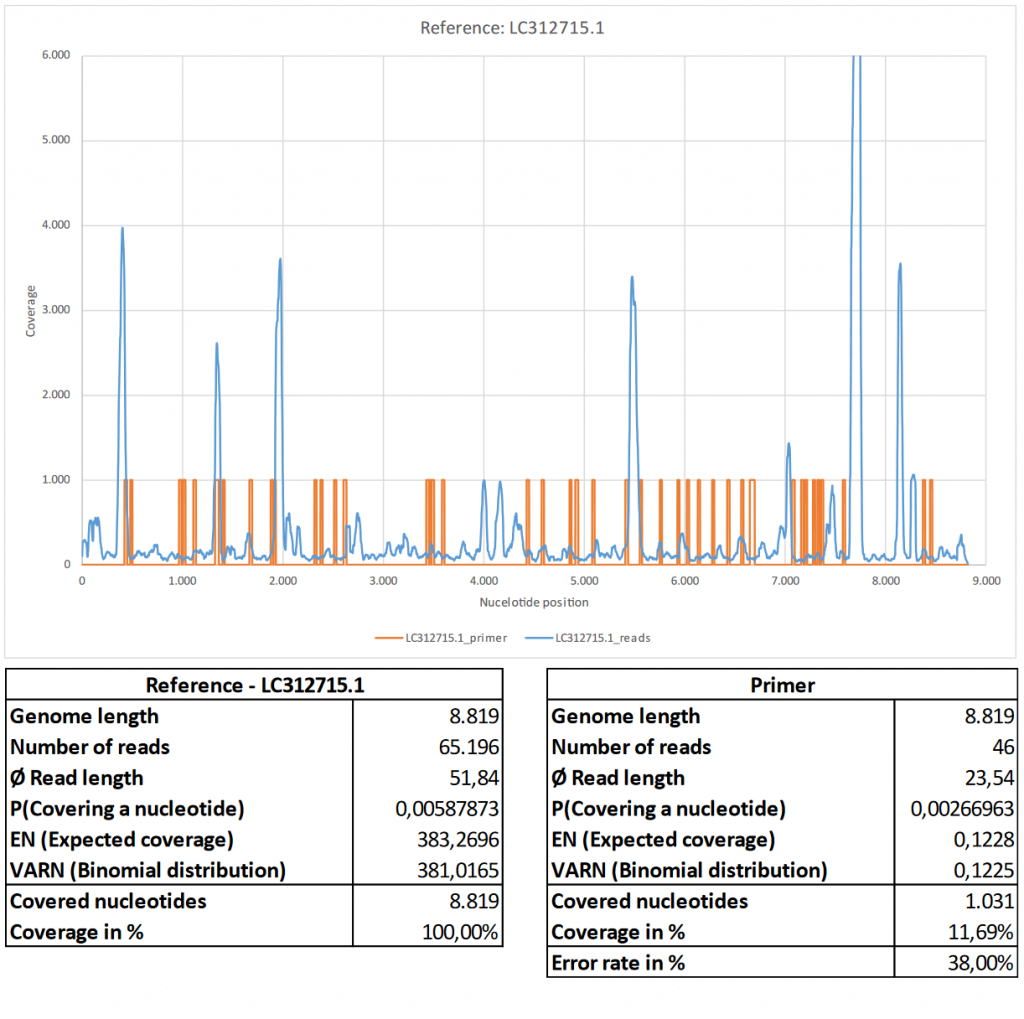
a) LC312715.1_short_reads cartographié en utilisant BBMap, (M1 ; M2) = (37 ; 0,60).
b) LC312715.1_primer cartographié en utilisant BBMap.
Déjà dans la section précédente, une grande similarité structurelle des séquences publiées avec la séquence de référence LC312715.1 a été montrée. La séquence consensus calculée a montré des taux d’erreur relativement plus faibles R1 = 8,60 % et R2 = 8,83 % en comparaison avec, par exemple, les références associées au SRAS. La figure 6 montre de nettes différences avec la figure 13. La distribution de la couverture montre également plus un modèle en forme de vague avec des zones relativement régulières de couverture particulièrement élevée et est donc clairement différente de la distribution de la couverture de la référence aléatoire « rnd_uniform ». La distribution des longueurs de lecture (Figure 23 b), comparer également c)) diffère également de manière significative des distributions plus aléatoires et montre un nombre significatif de lectures cartographiables avec des longueurs allant jusqu’à environ 110 nt. La longueur moyenne des lectures de 51,84 nt est également plus élevée que pour « rnd_uniform », par exemple.
Une fois encore, il est intéressant de noter la position des séquences d’amorce par rapport aux zones de couverture nucléotidique élevée par rapport à la couverture moyenne. Au total, 46 des 52 séquences d’amorce ont pu être assignées à la référence considérée ici avec un taux d’erreur de 38,00 %. La figure 6 suggère que les lectures de séquences courtes associées à la référence LC312715.1 ont également été amplifiées lors de la confirmation par PCR, malgré le fait que les séquences d’amorce n’ont pu être assignées à la référence qu’avec un taux d’erreur relativement élevé.
Enfin, passons à la référence KJ410048.1 (virus de la rougeole).

a) KJ410048.1_short_reads cartographié à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60).
b) KJ410048.1_primer cartographié à l’aide de BBMap.
La distribution de la couverture diffère sensiblement de celle de la figure 6 et présente certaines similitudes avec la distribution des lectures de séquences associées pour « rnd_uniform », avec une variation moindre dans les zones de moindre couverture. La distribution des longueurs de lecture (Tableaux et Figures : Figure 24 d)) ainsi que la longueur de lecture moyenne de 42,38 sont comparables aux données de « rnd_unifom » et sont également corrélées avec des taux d’erreur relativement élevés F1=28,70 % et F2=28,79 %.
Discussion et perspectives
Nous avons examiné les données de séquence publiées (numéro d’accession BioProject PRJNA603194 dans la base de données NCBI Sequence Read Archive [SRA]) sur la séquence du génome de SARS-CoV-2 (GenBank : MN908947.3) en utilisant une approche bioinformatique simple. Les méthodes que nous avons utilisées ne sont pas spécifiques au SARS-CoV-2 et peuvent être appliquées à d’autres données de séquençage sans modifications particulières.
Tout d’abord, nous avons répété la génération de contigs avec Megahit (v.1.2.9) en utilisant les données de séquence disponibles et avons obtenu des résultats significativement différents par rapport aux représentations de [1]. En particulier, nous n’avons pas été en mesure de reproduire le contig le plus long avec une longueur de 30 474 nt, qui selon [1] comprenait presque tout le génome viral et a servi de base pour la conception des amorces. Au contraire, le contig le plus long que nous avons généré (29 802 nt) a montré une correspondance presque complète avec la référence MN908947.3. Par conséquent, les données de séquence publiées ne peuvent pas être les lectures courtes originales utilisées pour la génération des contigs. Ceci est à considérer comme extrêmement problématique dans le contexte des publications scientifiques, car de cette manière il n’est plus possible de vérifier les résultats publiés. La possibilité de vérifier les hypothèses scientifiques publiées est l’essence même de la science vivante.
Contrairement à ce qui a été rapporté dans [1], il se peut que nous ayons trouvé des contigs avec une couverture élevée associés à des acides ribonucléiques (ribosomiques) d’origine humaine. Il est donc possible que tous les acides nucléiques associés à l’homme n’aient pas été éliminés lors de la construction du SARS-CoV-2. En outre, aucune preuve de la présence d’acides nucléiques viraux dans l’échantillon du patient n’a été fournie et, par conséquent, il est possible que des fragments d’acides nucléiques humains ou non viraux aient été utilisés pour construire la séquence virale revendiquée MN908947.3 dans une large mesure sans être détectés. Cette possibilité devrait être exclue par des expériences de contrôle.
Dans toutes les publications sur les génomes de référence analysés dans cette étude, les preuves nécessaires sur l’origine exacte des fragments de séquence utilisés pour la construction n’étaient pas non plus fournies et les expériences de contrôle nécessaires n’étaient pas publiées.
Nous tenons à mentionner ici que des expériences de contrôle ont peut-être déjà été réalisées de nombreuses fois sans être remarquées, ce qui montre la possibilité de construire des génomes du SARS-CoV-2 à partir d’échantillons humains non infectieux. Par exemple, le séquençage du génome entier à partir d’échantillons dont la valeur de base du Ct est supérieure à 35 est rapporté dans [5] et [17]. Cela pourrait réfuter le modèle viral du SARS-CoV-2.
L’analyse des distributions de la couverture nucléotidique ainsi que des distributions de la longueur des lectures de séquence mappables pour les séquences de référence respectives conduit à l’hypothèse d’une possible amplification involontaire de lectures de séquence non associées au SARS-CoV-2. En outre, il faut envisager la possibilité de la génération accidentelle de séquences qui n’étaient pas présentes dans l’échantillon initial mais qui ont été générées uniquement par les conditions d’amplification, telles que les séquences d’amorces utilisées et les cycles effectués. Cette possibilité nécessite donc la réalisation d’expériences de contrôle appropriées.
En plus de tenter de reproduire l’assemblage publié dans [1] avec les lectures de séquences publiées, nous avons envisagé une approche simple pour analyser la structure interne de grands ensembles de données de lectures de séquences courtes. Avec les données de séquence disponibles, nous avons pu calculer des séquences consensus pour les génomes de référence LC312715.1 (VIH) et NC_001653.2 (virus de l’hépatite delta) avec une plus grande précision que pour les séquences de référence que nous avons considérées comme associées aux coronavirus. Cela était particulièrement vrai pour la séquence bat-SL-CoVZC45 (GenBank : MG772933.1), qui a conduit à l’hypothèse d’origine du SARS-CoV-2. Ainsi, nous avons pu étayer notre hypothèse selon laquelle les séquences génomiques virales revendiquées sont des interprétations erronées dans le sens où elles ont été ou sont construites sans que cela soit remarqué à partir de fragments d’acides nucléiques non viraux. En particulier, nos résultats soulignent l’urgente nécessité de réaliser des expériences de contrôle appropriées. Pour chaque séquence génomique virale pathogène suspectée, un protocole évident serait de tenter d’assembler les séquences génomiques d’échantillons non suspectés correspondants en utilisant des protocoles identiques.
Nous avons observé des taux d’erreur R1 et R2 élevés dans les génomes de référence pour la rougeole, Ebola ou Marburg, où les fragments d’acide nucléique utilisés pour la construction ont été propagés dans des cellules Vero. La question de savoir si cela est dû aux sources d’acide nucléique elles-mêmes, aux conditions d’amplification utilisées (par exemple, les séquences d’amorces et le nombre de cycles) ou aux protocoles de séquençage (par exemple, les polymérases et les transcriptases inverses utilisées) reste ouverte.
En ce qui concerne nos résultats, outre la publication des données de séquence finales utilisées, nous recommandons toujours de publier les données de séquence résultant uniquement de l’amplification avec des hexamères aléatoires et des nombres de cycles modérés afin de fournir les données les plus impartiales possibles pour l’analyse structurelle.
Matériel et méthodes
Profondeur de couverture d’une séquence de référence avec des lectures de séquences courtes
Soit 𝐺 la longueur de la séquence de référence, Ø𝐿 la longueur moyenne de lecture, 𝑛 le nombre de lectures de séquences courtes, et 𝑁 la profondeur moyenne aléatoire de couverture de la séquence de référence avec les lectures de séquences courtes. Alors

L’expression Ø𝐿/𝐺 peut être considérée comme la probabilité de couverture d’un nucléotide dans la séquence de référence avec une lecture de séquence courte.
Génération de séquences de référence aléatoires
Le théorème suivant permet la simulation d’une variable aléatoire avec une fonction de distribution cumulative.
Théorème (principe d’inversion) [28]. Soit 𝑈 une variable aléatoire également distribuée sur l’intervalle (0,1). Soit 𝑋 une variable aléatoire avec une fonction de distribution cumulative 𝐹, et soit

Alors s’applique
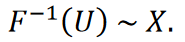
Soit 𝑈𝑖,𝑖 = 1, … ,29,903 des variables aléatoires équidistantes indépendamment identiques sur l’intervalle (0,1). Soit 𝑝𝑛𝑡,𝑛𝑡 ∈{𝐴,𝑇,𝐶,𝐺} la probabilité pour le nucleotide 𝑛𝑡. Ensuite, le nucléotide 𝑁𝑖,𝑖 = 1, … ,29.903 de la séquence de référence générée de façon aléatoire est obtenu via
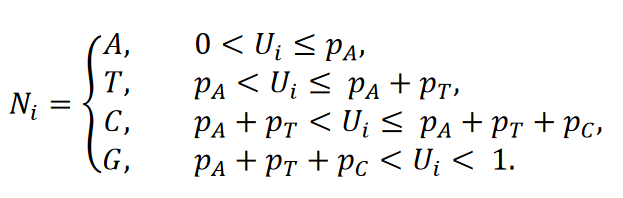
Pour la séquence de référence « rnd_unifom », la distribution uniforme sur l’ensemble {𝐴,𝑇,𝐶,𝐺} a été utilisée. Pour simuler la séquence de référence aléatoire “rnd_wuhan”, l’occurrence relative des nucléotides A, T, C et G dans la séquence du génome du SARS-CoV-2 (GenBank : MN908947.3) a été choisie comme distribution des nucléotides. Dans la construction des séquences de référence randomisées “rnd_wh_mk_1” et “rnd_wh_mk_2”, la probabilité conditionnelle, respectivement sur le dernier et sur les deux derniers nucléotides, a été choisie en fonction des fréquences empiriques correspondantes dans la séquence du SARS-CoV-2 (GenBank : MN908947.3).
Simulation stochastique de couvertures aléatoires d’une séquence de référence
La fonction de distribution cumulative de la distribution exponentielle avec le paramètre 𝜆 est [28],

Soit 𝑋 une variable aléatoire avec une fonction de distribution 𝐹. Alors 𝐸𝑋 = 1/𝜆 und 𝑉𝐴𝑅𝑋 = 1/𝜆2.
Méthodes bioinformatiques (analyse structurelle)
- Cartographie à l’aide de BBMap
bbmap.sh ref=$ reference. fasta
mapPacBio.sh in=SRR10971381_1.fastq in2=SRR10971381_2.fastq outm=mapped.sam vslow k=8 maxindel=0 minratio=0.1
- Sélection des séquences cartographiées en fonction de M1 et M2 en utilisant BBMap (reformat.sh)
reformat.sh in=mapped.sam out=sample_selection.sam
minlength=$ M1 (maxlength=100) idfilter=$ M2 ow=t
- Calcul de la séquence consensus
- 3.1. Préparation à l’aide de Samtools
samtools view — b sample_selection.sam > sample.bam
samtools sort sample.bam — o sample_sort_reads.bam
samtools index sample_sort_reads.bam - 3.2. Détermination de la séquence consensus préliminaire
samtools mpileup — uf mapping/$ reference. fasta
sample_sort_reads.bam | bcftools call -c | vcfutils.pl vcf2fq > SAMPLE_cns.fastq - 3.3. Détermination de la séquence consensus finale (min. Q20)
seqtk seq -aQ64 -q20 -n N échantillon_cns.fastq > échantillon_cns.fasta
- 3.1. Préparation à l’aide de Samtools
- Mappage de la séquence consensus à la séquence de référence en utilisant BWA.
bwa index $ reference. fasta
bwa mem $ reference. fasta sample_cns.fasta > sample_cns.sam
- Examen avec Tablet et Excel
L’évaluation a été réalisée à l’aide du logiciel Tablet pour la visualisation des données de séquence et du programme de feuille de calcul Excel.
Références
⚠ Les points de vue exprimés dans l’article ne sont pas nécessairement partagés par les (autres) auteurs et contributeurs du site Nouveau Monde.


















